Category: Ataxia
Objective: To describe a case series of HA patients from Chiloé, identifying genetic variants and analyzing phenotypes.
Background: The Chiloé Islands, located in southern Chile, constitute a geographically and genetically isolated region of approximately 166,000 inhabitants, characterized by high rates of consanguinity. Hereditary ataxias (HAs) are rare neurodegenerative disorders with estimated prevalence rates of 0.31 to 41 per 100,000 (1). Their genetic heterogeneity complicates diagnosis. Previous Chilean data identified spinocerebellar ataxia type 3 (SCA3) and Friedreich’s ataxia as the most common autosomal dominant (AD) and autosomal recessive (AR) forms, respectively (2).
Method: We evaluated 13 Chiloé patients diagnosed with HA. Clinical assessments included detailed neurological and family histories, the Scale for the Assessment and Rating of Ataxia (SARA) and the Montreal Cognitive Assessment (MoCA). Diagnostic evaluations included brain CT/MRI, electromyography and laboratory tests. Genetic analyses involved screening for common expansions associated with HA (SCA1, SCA2, SCA3) and whole-exome sequencing. RFC1 and FGF14 expansions were assessed in cases with suggestive phenotypes. Ethical approval was granted by the local IRB.
Results: The study included 13 individuals aged from 16 to 77 years. Mean age at disease onset was 45 ± 14.3 years with an average disease duration of 11 ± 6.7 years. Accompanying symptoms included polyneuropathy (46%), spasticity (38%) and cognitive impairment (16%). AD inheritance patterns were suggested in 62% of cases and AR in 38%. Genetic testing identified disease-causing variants in only 15% (one homozygous SPG7 and one RFC1 expansion), two individuals had a heterozygous likely pathogenic variant in SPG7 while 70% harbored variants of uncertain significance (VUS). A total of 21 VUS were identified in 10 different genes. Detailed clinical and molecular findings are listed in Tables 1 and 2
Conclusion: This study underscores the low diagnostic yield of conventional genetic testing in HA patients from isolated, underrepresented populations (URP). Common genetic causes were notably absent; most patients carried VUS, reflecting increased frequency in URP and emphasizing the critical need for diversifying genomic databases (3). The implementation of advanced genomic technologies, such as long-read sequencing, is recommended to enhance genetic diagnosis in genetically diverse and isolated URP (4).
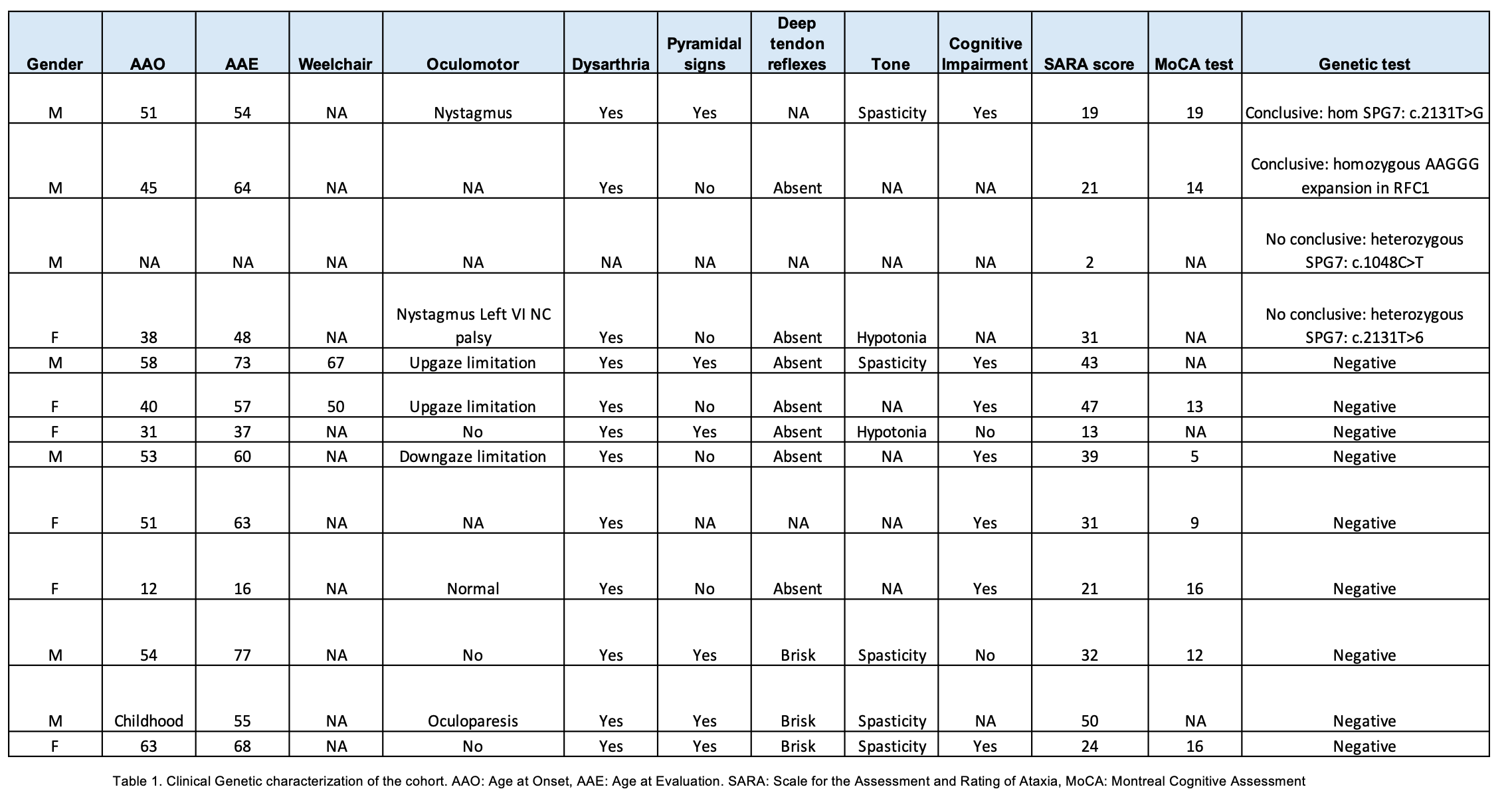
Table 1. Clinical Genetic characterization
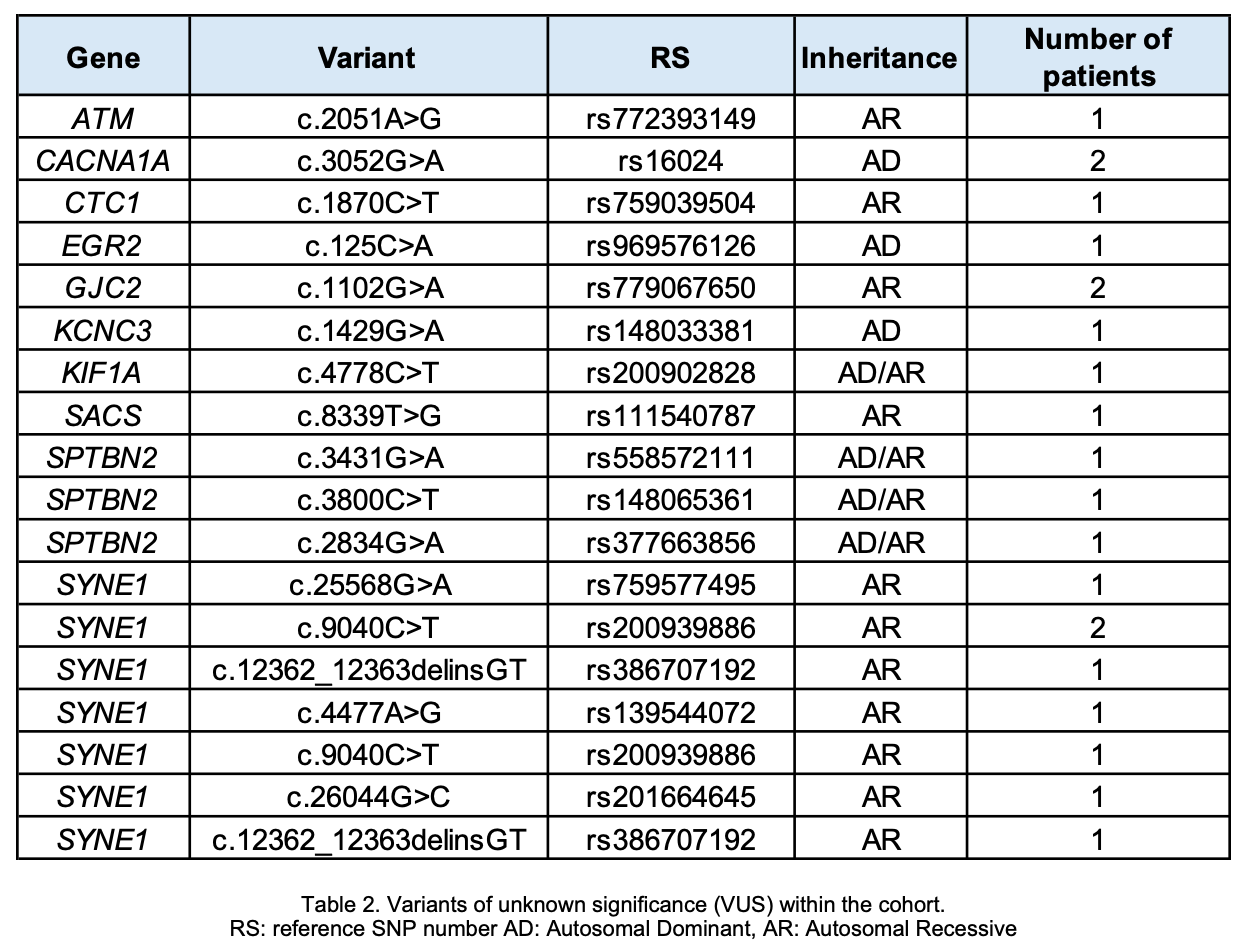
Table 2. Variants of unknown significance (VUS)
References: 1. Akbar U, Ashizawa T. Ataxia. Neurol Clin 2015; 33 (1): 225-48.
2. Saffie Awad, P., Vial Undurraga, F., & Chaná-Cuevas, P. (2018). Características clínicas de 63 pacientes con ataxia. Revista médica de Chile, 146(6), 702-707.
3. Chen, Elaine et al. “Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing.” JAMA network open vol. 6,10 e2339571. 2 Oct. 2023
4. Pellerin D, et al. Recent Advances in the Genetics of Ataxias: An Update on Novel Autosomal Dominant Repeat Expansions. Curr Neurol Neurosci Rep. 2025 Jan 16;25(1):16
To cite this abstract in AMA style:
E. Fernandez-Toledo, HM. Chaparro-Solano, P. Saffie-Awad. Case Series of 13 Hereditary Ataxia Patients From The Chiloe Islands, Chile. [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/case-series-of-13-hereditary-ataxia-patients-from-the-chiloe-islands-chile/. Accessed July 10, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/case-series-of-13-hereditary-ataxia-patients-from-the-chiloe-islands-chile/