Category: Myoclonus/Tics/Stereotypies
Objective: Mutations in the GOSR2 gene are associated with North Sea-Progressive Myoclonus Epilepsy (NS-PME). Because more recently additional phenotypes have been described we systematically reviewed all reported GOSR2 mutations, associated clinical phenotypes, and pathophysiological findings.
Background: NS-PME is a progressive neurological disorder caused by homozygous GOSR2 mutations (c.430G>T; p.Gly144Trp). Clinical symptoms include untreatable early-onset ataxia, cortical myoclonus, and epilepsy with continuous myoclonic jerks as major disabling symptom. Recently, the spectrum of GOSR2 mutations and associated phenotypes has expanded. Despite the increased clinical and genetic knowledge, the underlying pathophysiological mechanism caused by mutations in GOSR2 still remain largely elusive.
Method: A narrative review literature search was conducted in PubMed, EMBASE, and Web of Science (1985–August 2024) using the keywords ‘GOSR2’, ‘GS27 protein’, ‘Bos1’, and ‘Membrin’. Only studies in English and specifically studies on GOSR2 function, pathogenic variants, clinical manifestations, and potential therapies were included.
Results: A total of 42 patients with 11 different GOSR2 mutations were identified. Three main phenotypes were observed: 1. Progressive myoclonus ataxia/epilepsy (PMA/PME; n=31) 2. congenital muscular dystrophy (n=6) and 3. hearing loss (n=5). Additionally, orthopedic abnormalities (n=28) and endocrine dysfunction (n=8) were reported. Intercurrent infections or fever was a trigger of myoclonus/seizures in the PMA/PME phenotype and hearing loss phenotype.
In vitro and in vivo models show that GOSR2 mutations result in a (partial) loss of function of the GOSR2-binding SNARE complex. We show that specific mutations differently affect GOSR2 protein structure, and together with the involvement of specific isoforms, contribute to phenotypic variability.
Conclusion: GOSR2 mutations lead to progressive neurological disorders. The main phenotype is PMA/PME, and the majority of cases involves the c.430G > T; p. Gly144Trp founder variant. Mutations in GOSR2 lead to altered functional- and structural changes of the Golgi SNARE complex, impairing intracellular protein transport. Understanding how these alterations lead to the clinical spectrum of GOSR2-related diseases, will facilitate disease recognition, prognosis prediction and the development of more targeted treatment strategies.
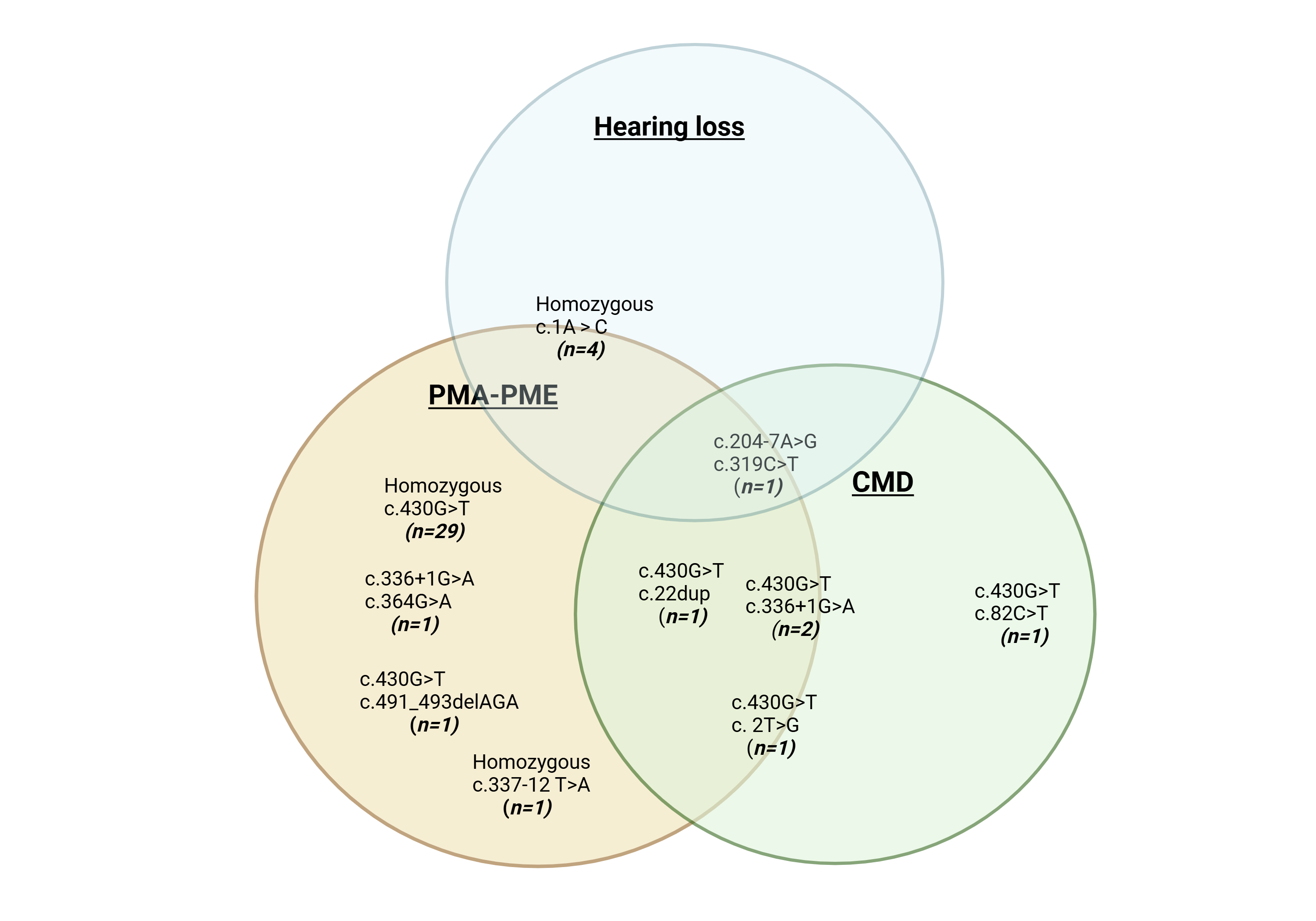
Phenotypes of GOSR2 mutations
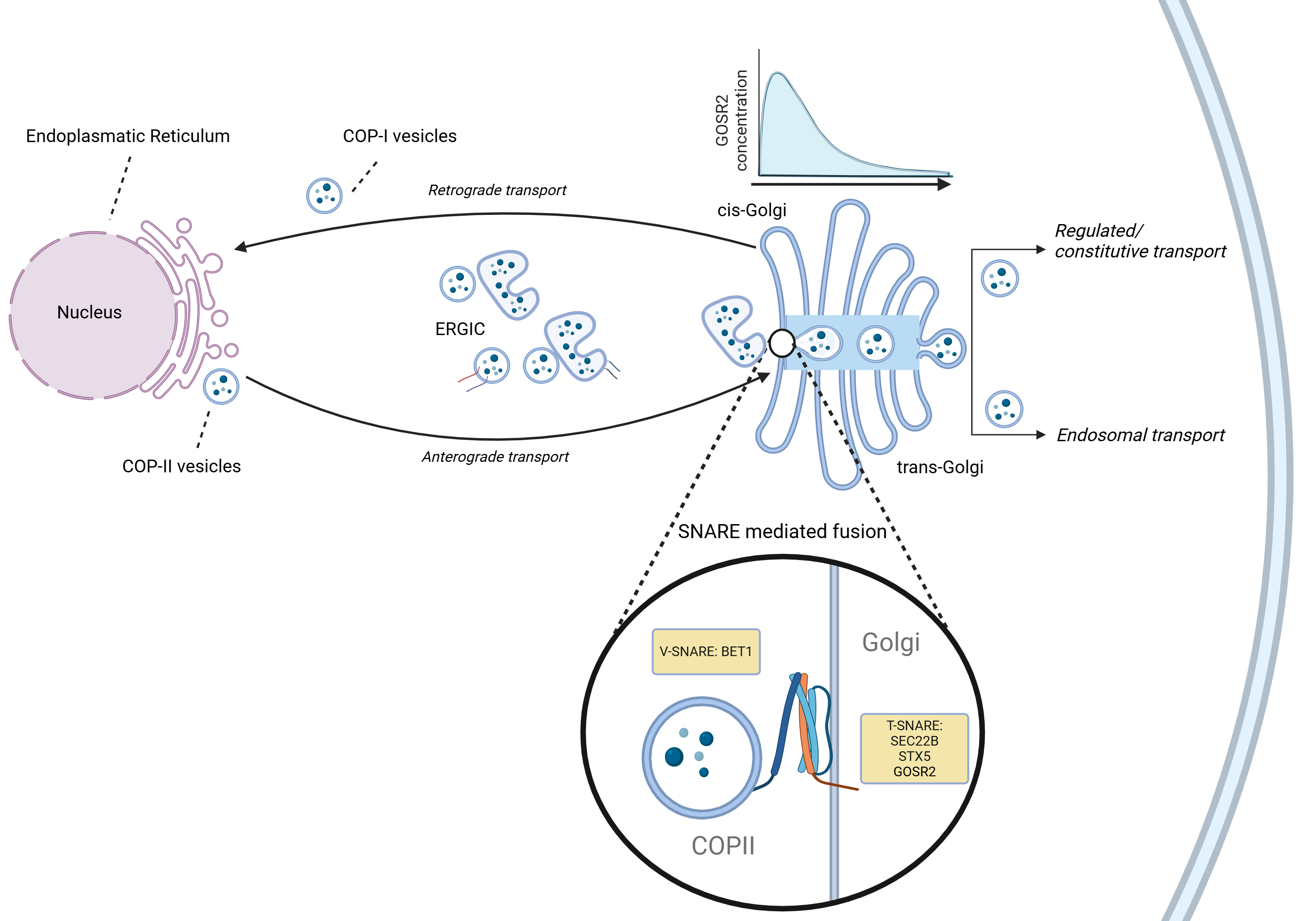
Function of GOSR2 as SNARE protein
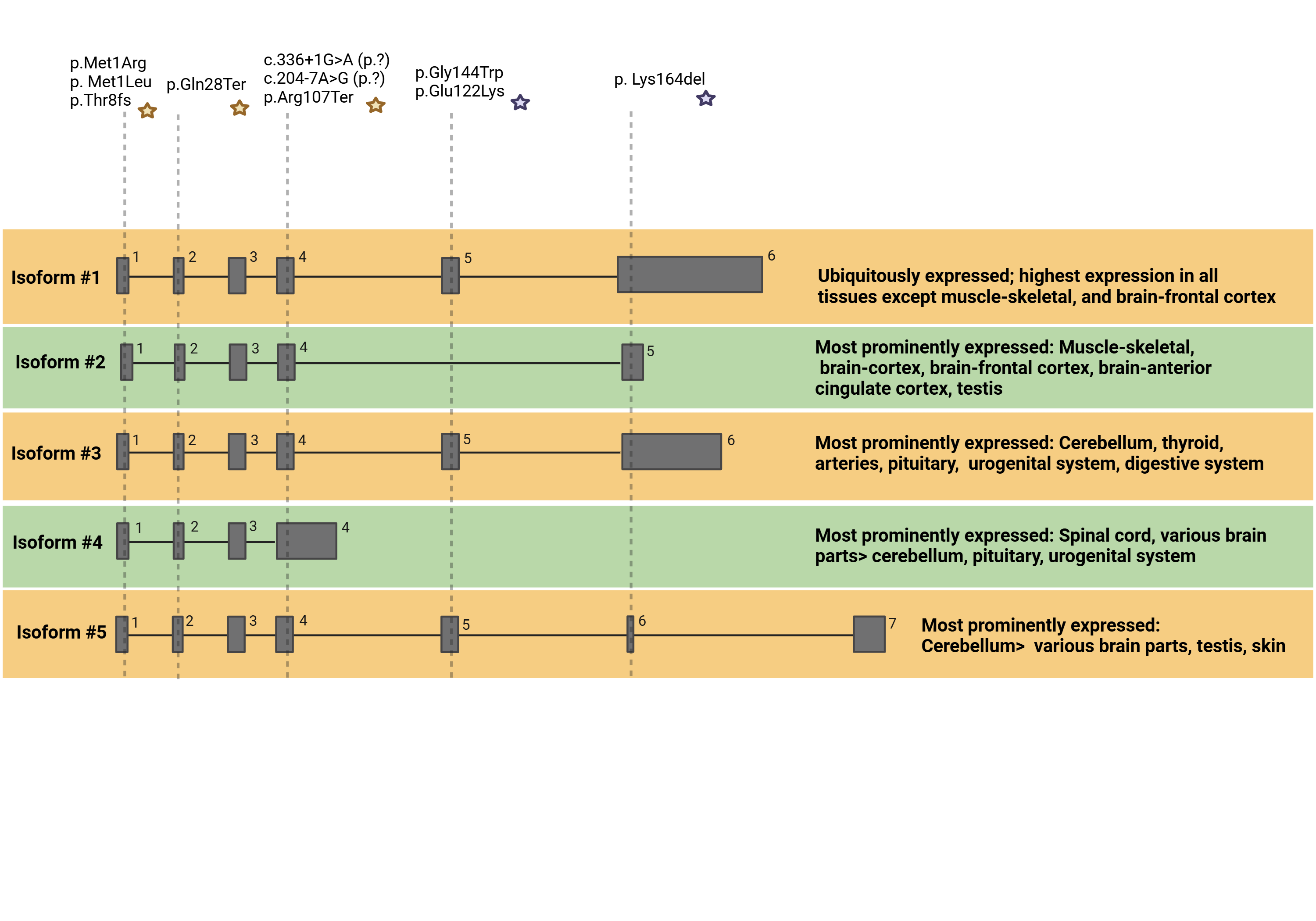
Isoforms of GOSR2
References: Figures are created in Biorender.
To cite this abstract in AMA style:
S. Polet, L. Siegal, S. Fuchs, M. Tijssen, T. de Koning. The Genotypic and Phenotypic Spectrum of GOSR2 Mutations: Clinical and Pathophysiological Insights [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/the-genotypic-and-phenotypic-spectrum-of-gosr2-mutations-clinical-and-pathophysiological-insights/. Accessed July 10, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/the-genotypic-and-phenotypic-spectrum-of-gosr2-mutations-clinical-and-pathophysiological-insights/