Category: Choreas (Non-Huntington's Disease)
Objective: To study the clinicoradiological and genetic profile of confirmed Dentatorubral–Pallidoluysian Atrophy (DRPLA) cases in Western India
Background: DRPLA is a hereditary disease caused due to trinucleotide repeat expansions in the ATN 1 gene with an autosomal dominant mode of Inheritance. It is rarely reported in the non-Japanese population. Here we present clinic-radiological features of nine DRPLA cases.
Method: A retrospective analysis of nine genetically confirmed DRPLA patients was conducted. Clinical data, including age at onset, initial symptoms, disease progression, family history, and neurological findings, were collected. Brain MRI was performed in all cases, and genetic testing for ATN1 CAG repeat expansions was conducted in seven patients. Patients were categorized based on the severity of their symptoms.
Results: The case series includes 8 male and 1 female patients. Their age of onset ranged from 8 to 55 years. 3 paediatric-onset cases (Cases 5, 6, 7) presented with, cortical myoclonus, generalised epilepsy and ataxia respectively. 5 adult-onset cases had varied presentation such as tremors, dystonia, cerebellar ataxia, cortical myoclonus or epilepsy which was well-controlled on anti-seizure medications. Cortical myoclonus and dementia were the most common findings, present in 6 patients, followed by epilepsy, which was present in 5 patients. 8 out of 9 patients had a positive family history of a hyperkinetic movement disorder, epilepsy or dementia. MRI brain was normal in 4 patients, 4 patients had cerebellar, cortical or pontine atrophy and one patient had diffuse B/L T2, FLAIR white matter hyperintensities. 7 out of 9 patients had abnormally increased CAG repeats in either allele of the ATN1 gene, and remaining 2 patients had a first degree relative with genetically confirmed DRPLA.
Conclusion: DRPLA manifests with severe phenotypic variability ranging from a severe chorea, ataxia and dementia to a milder myoclonic epilepsy phenotype with or without a positive family history. MRI findings also range from bilateral white matter hyperintensities to a normal MRI. Due to wide phenotypic variability, early diagnosis of DRPLA becomes challenging. In our patients number of CAG repeats and disease severity does not show any corelation. DRPLA was considered a rare entity outside the Japanese population, however rising number of cases reported from India and all across the world suggests the contrary.
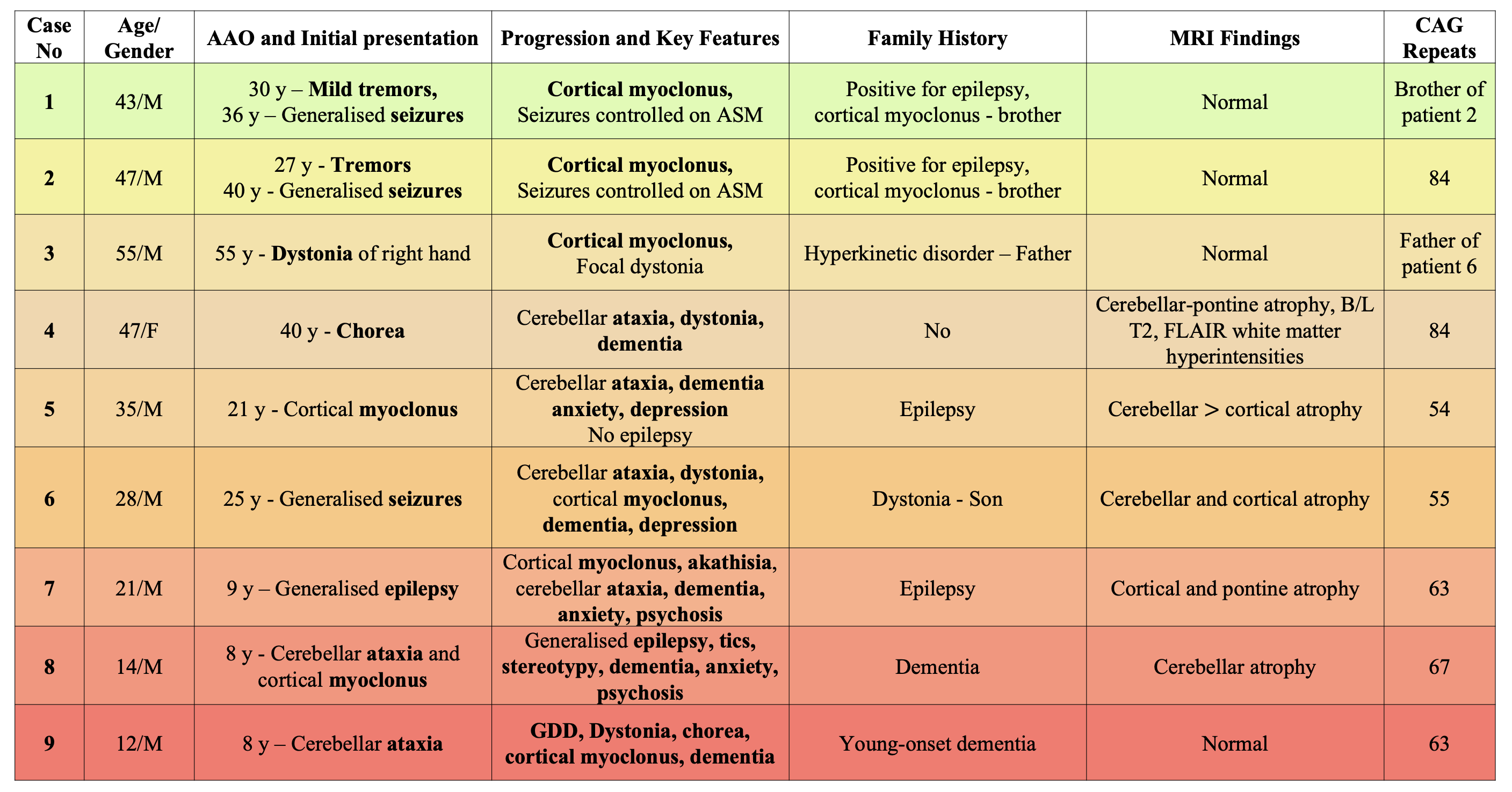
DRPLA cases from western India
To cite this abstract in AMA style:
K. Bavdhankar, P. Agarwal, N. Jain, S. Kothari, A. Soni, S. Kharat, A. Shah, S. Jagtap. Dentatorubral–Pallidoluysian Atrophy (DRPLA) : A case series of nine patients from western India [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/dentatorubral-pallidoluysian-atrophy-drpla-a-case-series-of-nine-patients-from-western-india/. Accessed April 10, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/dentatorubral-pallidoluysian-atrophy-drpla-a-case-series-of-nine-patients-from-western-india/