Objective: Identify proteomic and metabolomic biomarkers for Parkinson’s Disease (PD) progression diagnosis using statistical and machine learning (ML) approaches, leveraging data from cerebrospinal fluid (CSF) and plasma.
Background: PD is a prevalent movement disorder. Current diagnosis depends on late-manifesting motor symptoms. Analytic advancements allow deeper multi-omics data analysis for early diagnosis and disease classification. This study integrates proteomic and metabolomic profiles to uncover biomarkers for improved monitoring.
Method: Using untargeted CSF and plasma data from the Parkinson’s Progression Markers Initiative (PPMI). Aiming to characterize the proteomic and metabolic landscape of PD and prodromal patients. A comparative analysis of ML models -regression, random forest (RF), support vector machines (SVM), XGBoost, and neural networks (NN)-to identify proteins with predictive value for disease classification. Comparative analysis on metabolomic data to identify disease-specific expression patterns
Results: 116 significant proteins in PD patients identified. ML modeling highlighted 27 proteins with high feature importance in distinguishing PD cases. Multinomial regression demonstrated highest accuracy (86% in CSF, 79% in plasma), followed closely by NN (77% in CSF, 73% in plasma). The 27 proteins were significant (P<0.05) when considering the interaction effect between biofluid type and cohort [Figure 1]. Pairwise analysis identified 124 plasma metabolites (P <0.05): 6 Control vs. PD, 22 Control vs. Prodromal, 96 PD vs. Prodromal, and 71 in CSF :13 Control vs. PD, 4 Control vs. Prodromal, 15 PD vs. Prodromal. Progression analysis revealed non-linear relationships across disease stages, with prodromal stage showing peaks or dips in metabolite expression [Figure 2]
Conclusion: Dopamine-related metabolites show a prodromal-specific pattern, suggesting potential compensatory mechanisms during disease progression. Five top candidate proteomic biomarkers – apolipoproteins (A-IV, C-III, A-II, E, and L1), suggest a role in PD pathogenesis. Gene Ontology linked 11 proteins to fibrinolysis and blood coagulation regulation. KEGG pathway analysis identified cholesterol metabolism, complement, and coagulation cascades, and chylomicron remodeling as significantly associated with model features (P < 0.05). Correlation analysis highlighted protein expression changes relevant to disease progression.
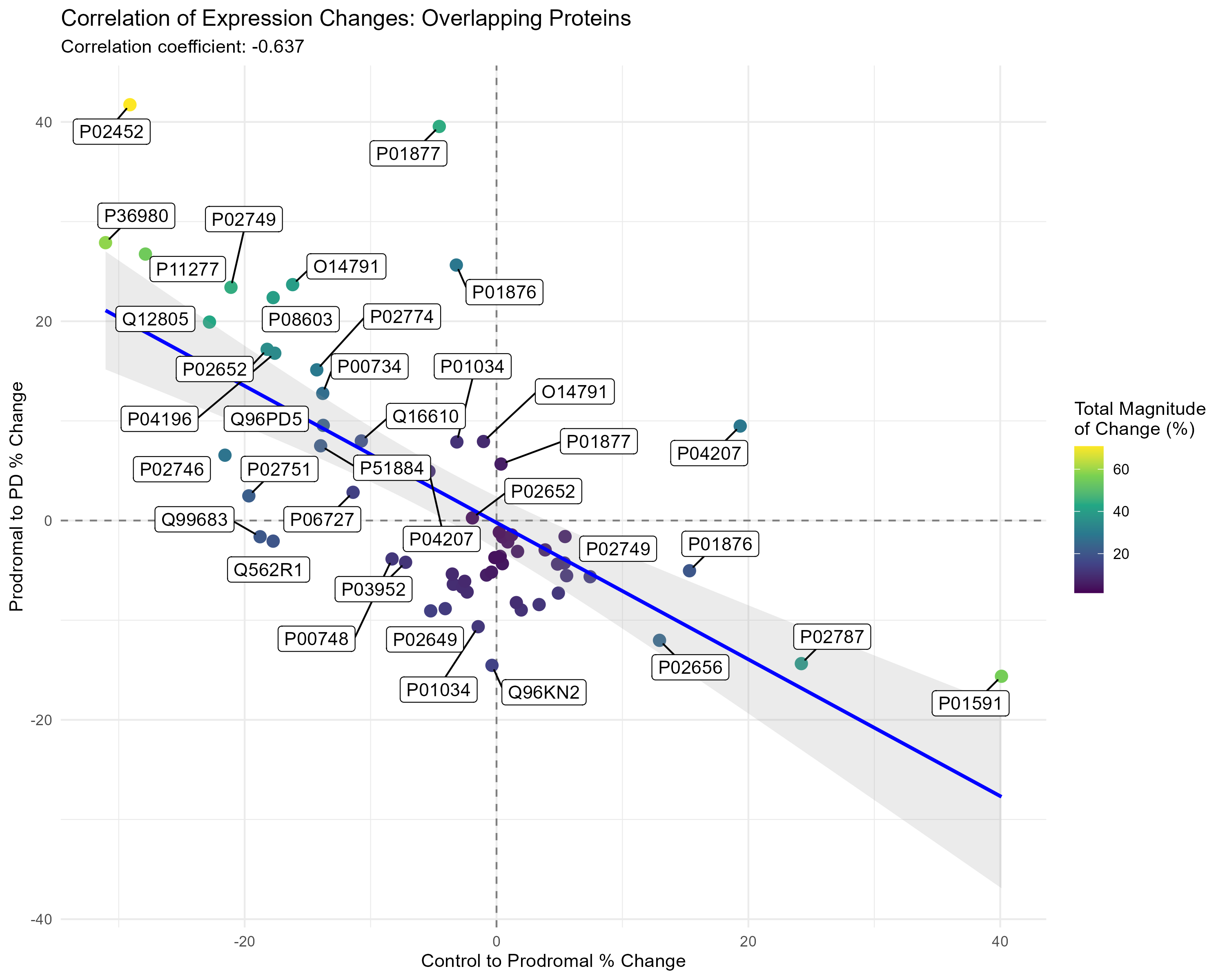
Figure 1- Protein Correlation Plot
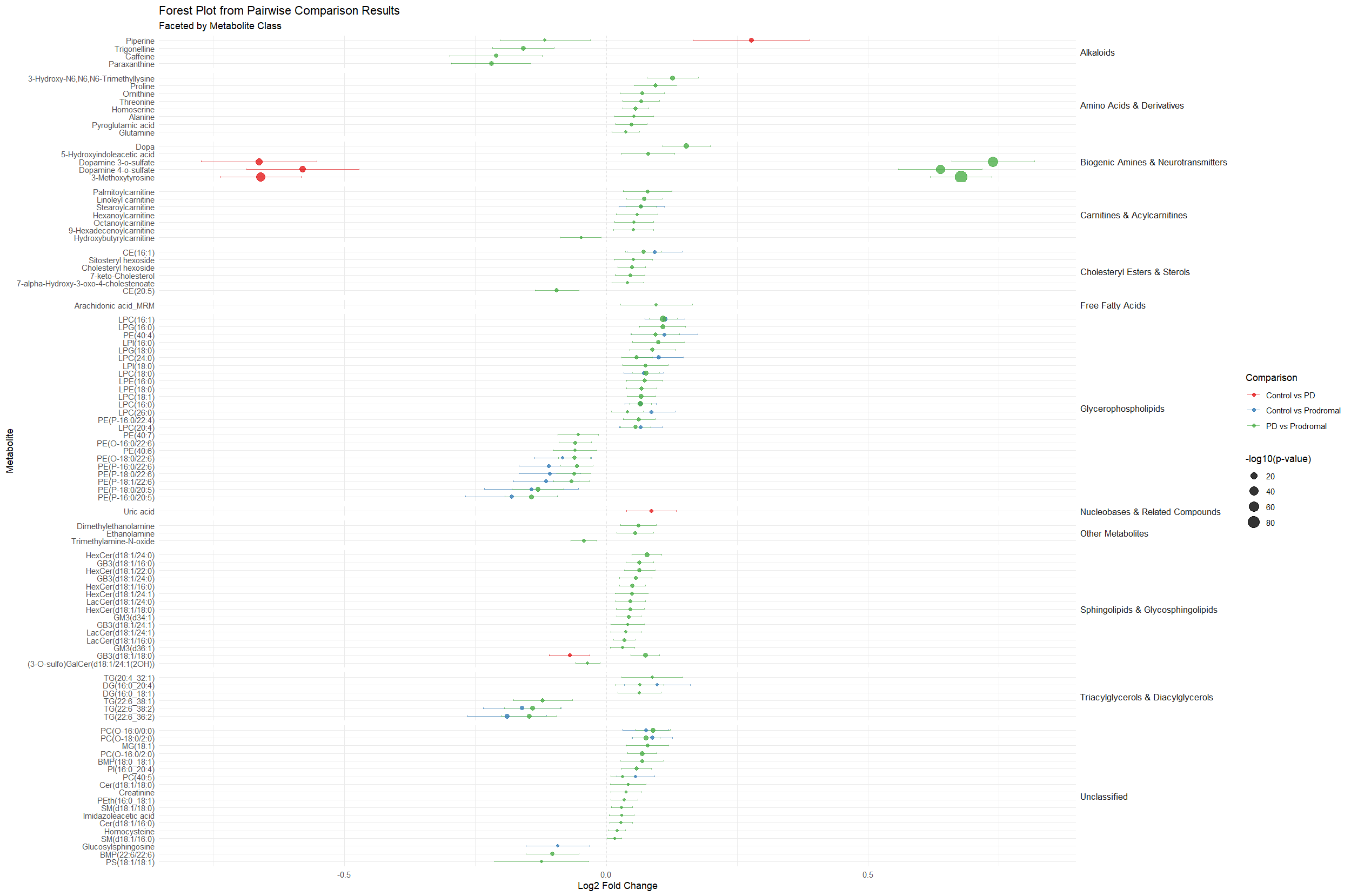
Fig 2a Plasma comparison and classification
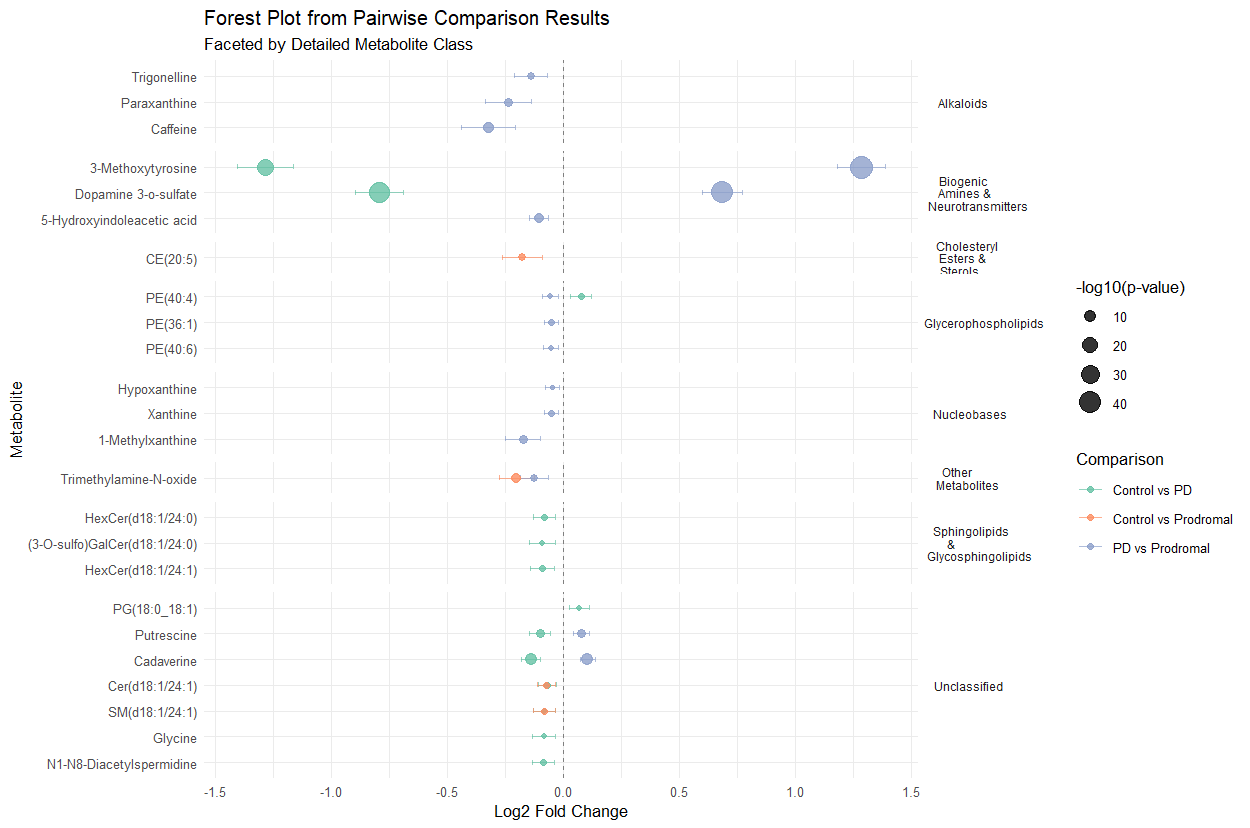
Fig 2b CSF comparison and classification
To cite this abstract in AMA style:
A. Galal, A. Moustafa, M. Salama. Identification of Metabolomic & Proteomic Panels for Parkinson’s Disease progression Diagnosis [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/identification-of-metabolomic-proteomic-panels-for-parkinsons-disease-progression-diagnosis/. Accessed April 6, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/identification-of-metabolomic-proteomic-panels-for-parkinsons-disease-progression-diagnosis/