Category: Ataxia
Objective: To report the first documented case of spinocerebellar ataxia type 17 (SCA17) in northern Mexico.
Background: SCA17 is an autosomal dominant neurodegenerative disorder caused by the expansion of the CAG/CAA trinucleotide coding for glutamine in TBP, located at 6q27 [1]. It is clinically characterized by cerebellar ataxia, accompanied by manifestations such as chorea, dystonia, parkinsonism, pyramidal signs, cognitive impairment, and psychiatric symptoms [2].
Worldwide, fewer than 100 families with SCA17 have been reported, while in Mexico, 3 families and 2 sporadic cases have been documented, making it the least prevalent among documented autosomal dominant cerebellar ataxias in the country [3,4].
Method: Case report.
Results: A 34-year-old male started at age 32 with dysarthria and gait lateralization, followed by dystonia affecting the face, neck, and left hemibody. Physical examination revealed visuoconstructive apraxia, ventromedial dysexecutive syndrome, emotional flattening, apathy, lack of motivation, cerebellar dysarthria, hypometric saccades, left laterocollis, left hemibody dystonia, global rigidity, dysmetria, dysdiadochokinesia, and ataxic gait [figure1].
His 60-year-old father started at age 52 with mild cognitive impairment, ataxia, and parkinsonism. His 15-year-old daughter has had cognitive deficits and ataxia since age 9, currently dependent on daily living activities.
Laboratory studies, including serum heavy metal levels, TSH, and peripheral blood smear, were unremarkable. Huntington’s disease was excluded with normal Huntingtin levels. Brain MRI showed cerebellar atrophy of both hemispheres and vermis, as well as corticosubcortical cerebral atrophy with ventricular dilation due to brain volume loss [figure2].
Due to family history, whole-genome sequencing was requested, revealing heterozygous CAG/CAA expansion in TBP (49-54 repeats), confirming the diagnosis of SCA17. Allelic triplet CAG/CAA expansion was evaluated in the patient’s daughter, detecting heterozygous expansion in TBP (57±1 repeats) within the full penetrance range for SCA17.
Conclusion: SCA17 should be considered when there is a family history of related symptoms. In our context, whole-genome sequencing is useful to differentiate between differential diagnoses, avoiding multiple costly specific tests for the patient. The diagnosis allows for medical counseling to improve the quality of life in these patients.
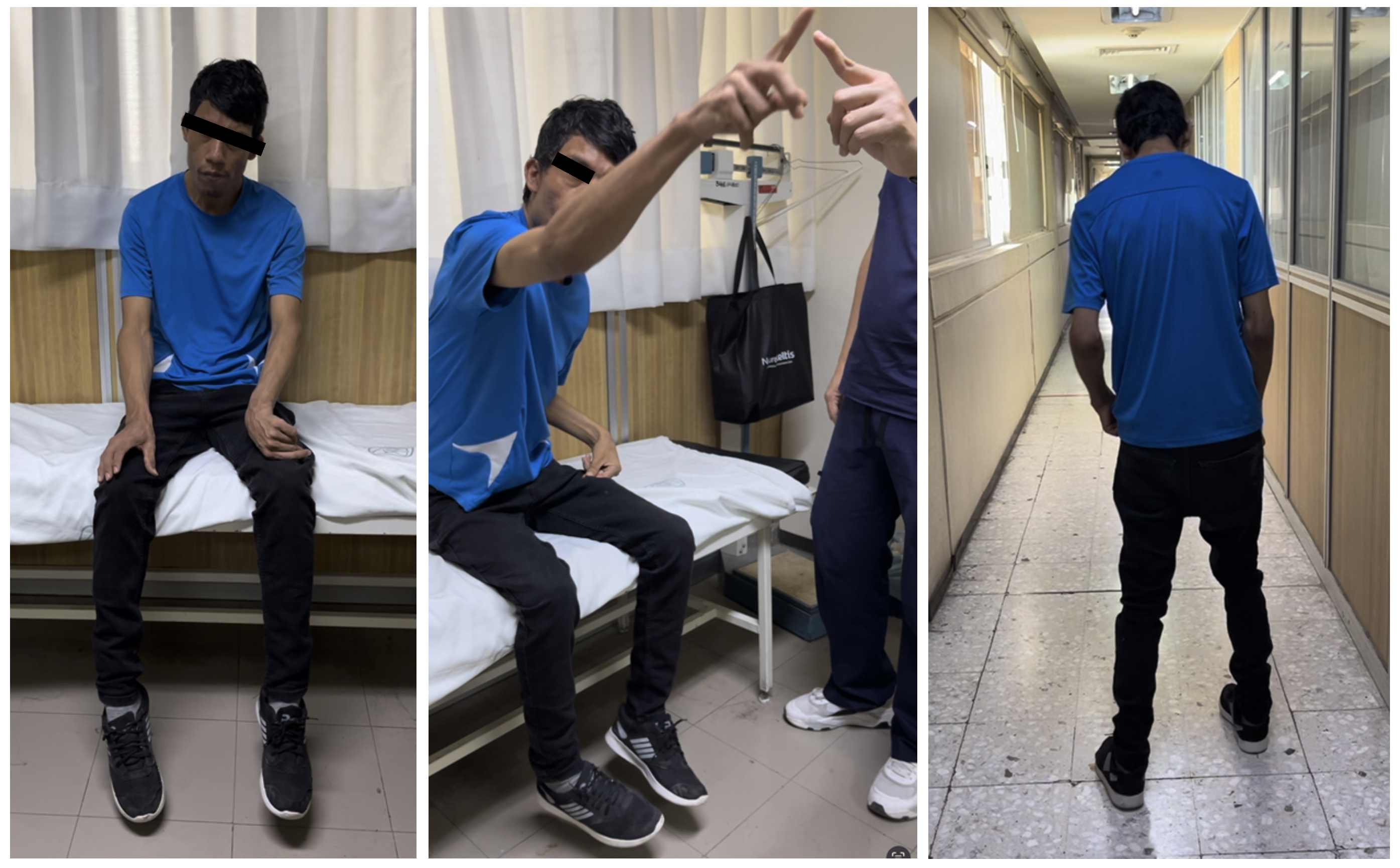
Figure1
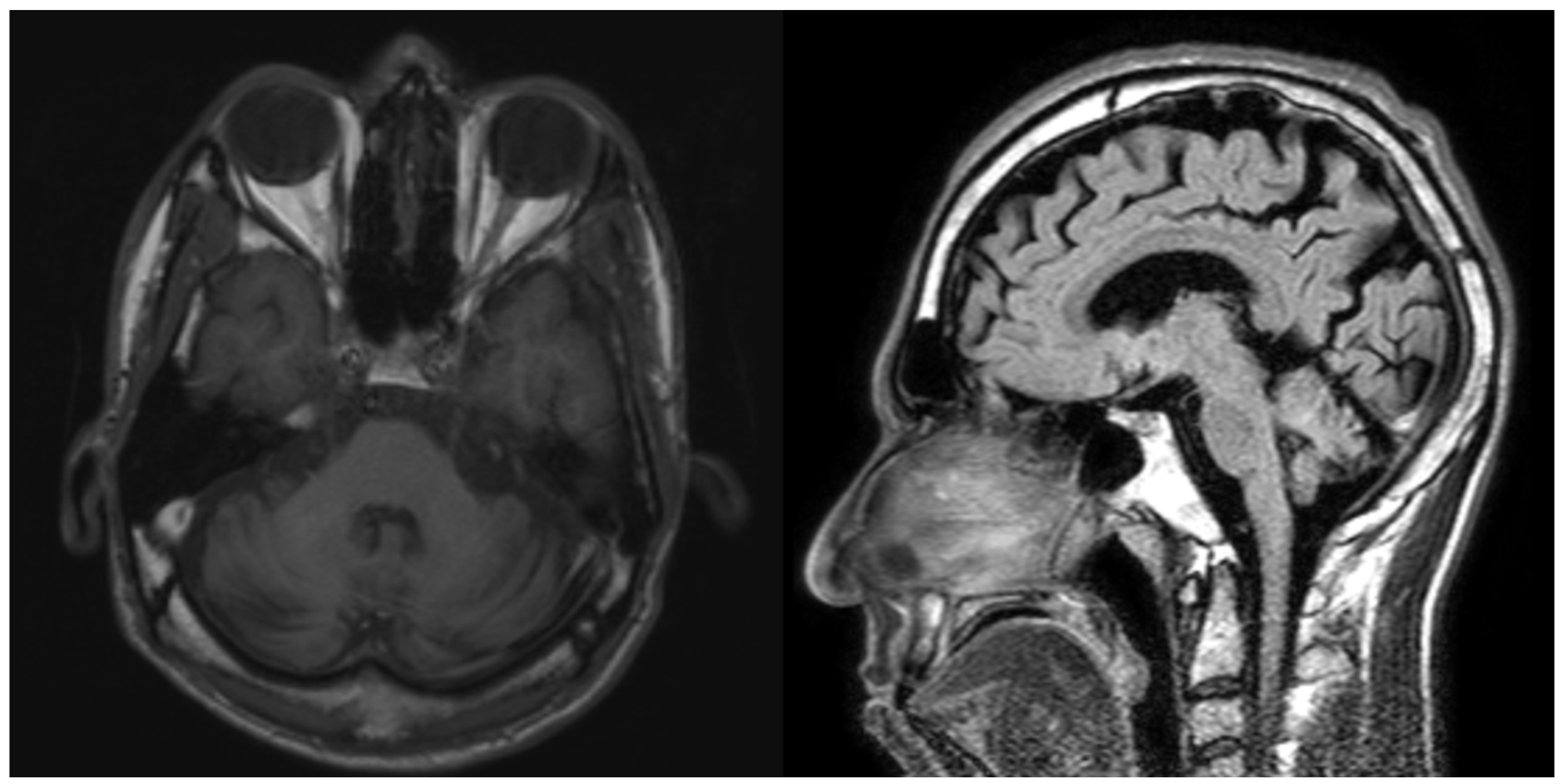
Figure2
References: 1. Origone P, Gotta F, Lamp M, Trevisan L, Geroldi A, Massucco D, et al (2018). Spinocerebellar ataxia 17: full phenotype in a 41 CAG/CAA repeats carrier. Cerebellum & Ataxias. 14;5(1):7.
2. Rossi, M., Hamed, M., Rodríguez-Antigüedad, J., Cornejo-Olivas, M., Breza, M., Lohmann, K., Klein, C., Rajalingam, R., Marras, C., & van de Warrenburg, B. P. (2023). Genotype-Phenotype Correlations for ATX-TBP (SCA17): MDSGene Systematic Review. Movement disorders : ficial journal of the Movement Disorder Society, 38(3), 368–377.
3. Toyoshima Y, Onodera O, Yamada M, et al. (2005, Updated 2022 Jul 28). Spinocerebellar Ataxia Type 17. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
4. Alonso E, Martínez‐Ruano L, de Biase I, Mader C, Ochoa A, Yescas P, et al. (2007). Distinct distribution of autosomal dominant spinocerebellar ataxia in the Mexican population. Movement Disorders Society; 15;22(7):1050–3.
To cite this abstract in AMA style:
S. Murillo Quintana, D. Sánchez Galván, J. Mejía Chávez, S. Flores Casas, L. Hernández Salomón. Spinocerebellar Ataxia Type 17 First Reported Case in Northern Mexico [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/spinocerebellar-ataxia-type-17-first-reported-case-in-northern-mexico/. Accessed April 7, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/spinocerebellar-ataxia-type-17-first-reported-case-in-northern-mexico/