Category: Ataxia
Objective: To present a case of two affected siblings with Spinocerebellar ataxia type 1
Background: Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant neurodegenerative disorder caused by CAG mutations in the ATXN1 gene, encoding ATXN1 protein with a pathogenic polyglutamine expansion. SCA1 patients typically have more than 39 CAG repeats on the affected allele [1-2]. SCA1 is characterized by cerebellar ataxia, bulbar dysfunction, and ocular manifestations such as hypermetric saccades, irregular pursuits and nystagmus. Extracerebellar signs, including cognitive decline and psychiatric symptoms, are common. Progressive respiratory failure is the primary cause of death. Treatment primarily focuses on supportive care to improve quality of life. Riluzole has been studied as a safe treatment for ataxia of various etiologies, but efficacy has been mixed in meta-analyses and placebo-controlled trials [3].
Method: We report the clinical and genetic testing results in two affected siblings with SCA1.
Results: Two patients in their early 40s presented with gait instability and dysarthria for 4-5 years. One developed dysphagia. Neurologic examination showed hypermetric saccades, scanning and monosyllabic speech, bilateral dysmetria and dysdiadochokinesia, as well as pyramidal signs. One patient could stand with assistance, while the other exhibited erratic gait with irregular speed, variable step length, and leftward veering. Brain imaging showed cerebellar volume loss exceeding age-related expectations. SARA scores during visits were 26 and 29. Family history revealed consanguinity (paternal first-cousin marriage) and Yemeni ethnicity. A pedigree demonstrated autosomal dominant inheritance [Figure 1]. Whole exome sequencing in one sibling identified 53 CAG repeats on the affected allele and 30 on the unaffected. Both were prescribed riluzole, physical therapy and speech therapy. SARA scores are monitored every 3-6 months to assess disease progression and treatment efficacy.
Conclusion: This case underscores the importance of recognition of SCA1 classic presentations and exam findings. Riluzole is likely safe and has shown promise in a mixed group of individuals including patients with SCA1, however further investigation is needed. Genetic testing and counselling play an important role when divulging and treating degenerative and fatal neurologic disease.
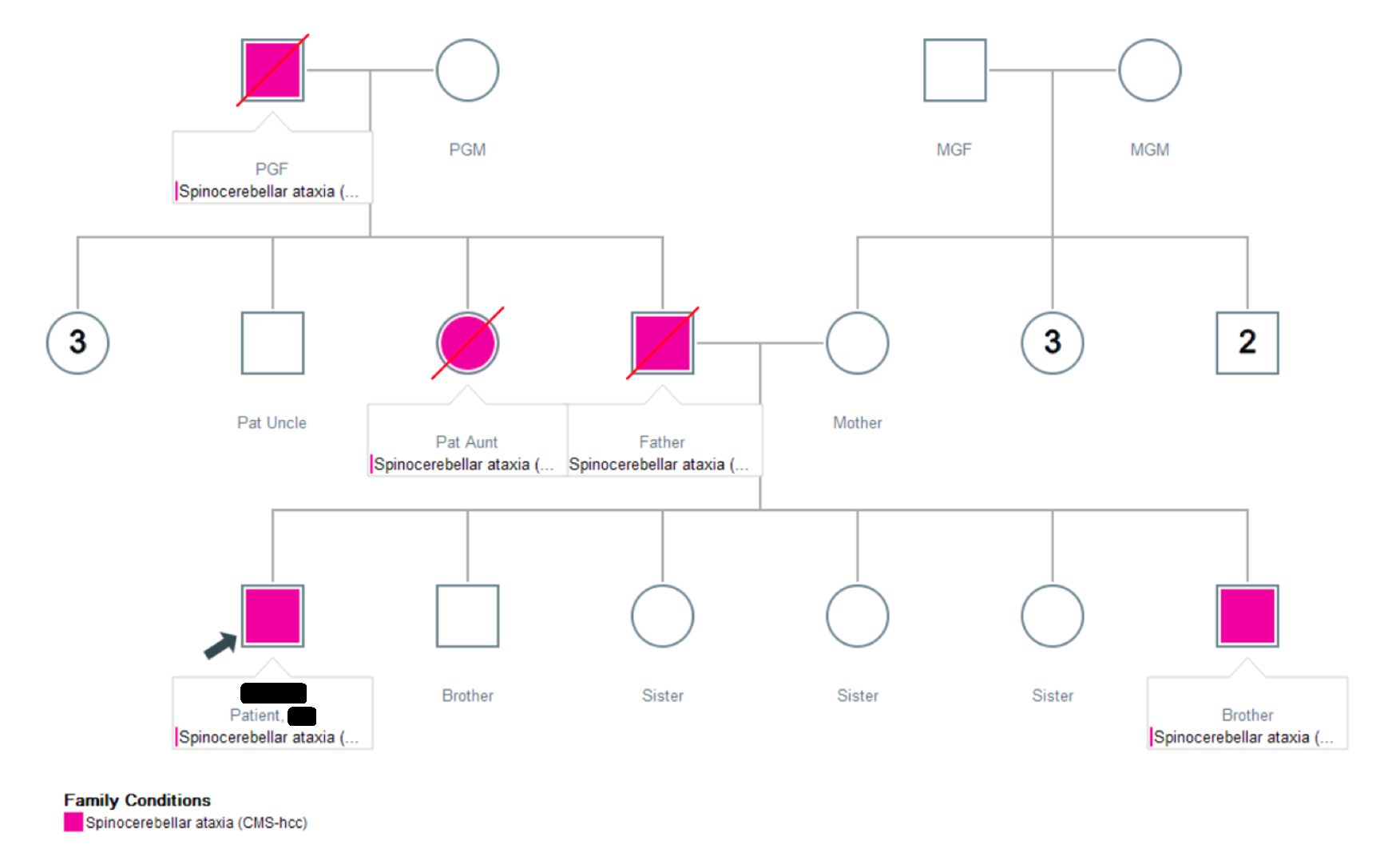
Pedigree of two patients with SCA1
References: [1] Donato SD, Mariotti C, Taroni F. Spinocerebellar ataxia type 1. Handb Clin Neurol. 2012;103:399-421. doi:10.1016/B978-0-444-51892-7.00025-5
[2] Opal P, Ashizawa T. Spinocerebellar Ataxia Type 1. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2025.
[3] Zesiewicz TA, Wilmot G, Kuo SH, et al. Comprehensive systematic review summary: Treatment of cerebellar motor dysfunction and ataxia: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018;90(10):464-471. doi:10.1212/WNL.0000000000005055
To cite this abstract in AMA style:
S. Fu, U. Agarwal, I. Goldszer. A Case Report of Two Affected Siblings with Spinocerebellar Ataxia Type 1 [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/a-case-report-of-two-affected-siblings-with-spinocerebellar-ataxia-type-1/. Accessed April 10, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/a-case-report-of-two-affected-siblings-with-spinocerebellar-ataxia-type-1/