Category: Choreas (Non-Huntington's Disease)
Objective: Here we describe for the first time two unrelated cases of genetically confirmed Choreoacanthocytosis from Algeria.
Background: Choreoacanthocytosis is a rare and severe inherited neurodegenerative cause of chorea linked to biallelic pathogenic variants in the VPS13A gene[1], [2]. It is characterized by a variable phenotype associating different degrees of chorea, parkinsonism, dystonia, gait instability, neuropathy, psychiatric symptoms and the presence of acanthocytes in the blood [3].
Method: Case report
Results: Case A: A 33 years old male born from a consanguineous marriage and having three sisters with the same symptomatology, one of them deceased [figure1]. He presented a five years history of irritability and progressively worsening generalized choreic movements and involuntary oro-facial and lingual dyskinesia. Upon examination he had a mild cognitive impairment (MMSE=24/30), generalized hypotonia and signs of lip biting and mutilation, Head drops were frequent and Brain MRI was unremarkable, creatine kinase levels were elevated (x8 normal value) and acanthocytes were present in blood smear. EMG identified axonal sensorimotor neuropathy.
Case B: a 31years old female presented for two years progressive involuntary choreic movements of the trunk and the limbs with frequent tongue protrusions and oro-facial dyskinesia. She presented also cervical dystonia, hypotonia and severe dysarthria. No consanguinity or similar cases in the family were reported [figure2]. MRI showed T2 Hypersignal in the caudat nuclei and putamens, Copper and ceruloplasmin blood levels were normal. Whole exome sequencing performed in both cases identified two known missens pathogenic homozygous variants in the VPS13A (NM_033305.3) gene; c.7214T>G (p.L2405T, rs919186738) in case A and c.2593C>T (p.R865T, rs766404788) in case B. Choreoacanthocytosis typically manifests in the third or fourth decade, head drops are pathognomonic but inconstant [4]. Blood smears are very useful simple tests that can allow early diagnosis but are also found in other Neuroacanthocytosis syndromes [3], [5].
Conclusion: Choreoacanthocytosis is a rare and severe disease that can be diagnosed clinically when having typical features, to this date no effective treatment is available, botulinum toxin and DBS are the main options to ease patients’ symptoms.
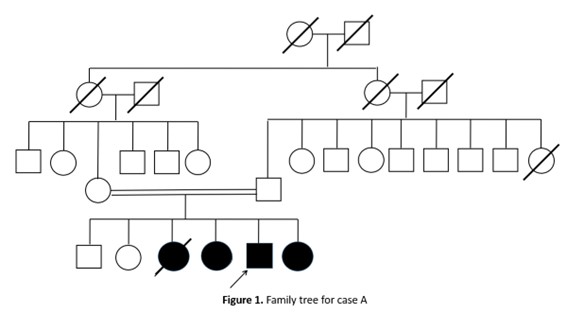
Figure 1. Family tree of case A
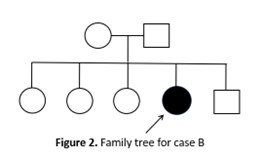
Figure 2. Family tree of case B
References: [1] L. Rampoldi, A. Danek, and A. P. Monaco, “Clinical features and molecular bases of neuroacanthocytosis,” J. Mol. Med. Berl. Ger., vol. 80, no. 8, pp. 475–491, Aug. 2002, doi: 10.1007/s00109-002-0349-z.
[2] S. Ueno et al., “The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis,” Nat. Genet., vol. 28, no. 2, pp. 121–122, Jun. 2001, doi: 10.1038/88825.
[3] K. Peikert, A. Danek, and A. Hermann, “Current state of knowledge in Chorea-Acanthocytosis as core Neuroacanthocytosis syndrome,” Eur. J. Med. Genet., vol. 61, no. 11, pp. 699–705, Nov. 2018, doi: 10.1016/j.ejmg.2017.12.007.
[4] S. A. Schneider, A. E. Lang, E. Moro, B. Bader, A. Danek, and K. P. Bhatia, “Characteristic head drops and axial extension in advanced chorea-acanthocytosis,” Mov. Disord., vol. 25, no. 10, pp. 1487–1491, 2010, doi: 10.1002/mds.23052.
[5] J. Feriante and V. Gupta, “Neuroacanthocytosis,” in StatPearls, Treasure Island (FL): StatPearls Publishing, 2024. Accessed: Mar. 15, 2024. [Online]. Available: http://www.ncbi.nlm.nih.gov/books/NBK560767/
To cite this abstract in AMA style:
Y. Mecheri, S. Talbi, A. Rezigue, M. Zouzou, BS. Fekraoui, F. Serradj, A. M'Zahem. Choreoacanthocytosis: The First Genetically Confirmed Cases From Algeria [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/choreoacanthocytosis-the-first-genetically-confirmed-cases-from-algeria/. Accessed July 4, 2026.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/choreoacanthocytosis-the-first-genetically-confirmed-cases-from-algeria/