Category: Rare Neurometabolic Diseases
Objective: To characterize the spectrum of neurological manifestations of Gaucher disease type 3 (GD3) and identify the most common GBA1 variants.
Background: Gaucher disease is an autosomal recessive lysosomal storage disorder caused by pathogenic biallelic variants in the GBA1 gene (NM_000157.3). It is classified into three main phenotypes, of which the chronic neuronopathic type (GD3) is characterized by progressive neurological involvement in infancy or childhood[1].
Method: Focusing on the group of GD3 patients, we analyzed data from a recently published MDSGene systematic review on genotype-phenotype relationships of GBA1 variants[2].
Results: Out of 226 publications, we obtained clinical and genetic data from 1.408 GD3 unique patients originating from 42 countries. Slightly more patients were male (53.1%), had a positive family history of GD (65.5%), a median age at onset of 1 year (IQR: 2 years, range: 0-23 years), and were of Asian (23.5%) and White (23.1%) ethnicities. The consanguinity rate was high (78.3%) and 75.7% of the patients carried a homozygous variant. The majority of 104 reported different variants were classified as “severe” GBA1 variants, of which p.Leu483Pro was the most common (77.0%), followed by p.Phe252Ile and p.Arg502Cys (3.5% each). The most frequent neurological manifestations were developmental delay (64/68, 94.1%), intellectual disability (93/149, 62.4%), seizures (136/218, 62.4%), saccadic eye abnormalities (146/147, 99.3%), and gaze palsy (151/159, 95.0%). The main associated movement disorders were ataxia (44/49, 89.8%) and myoclonus (65/75, 86.7%), often found in isolation rather than combined with other movement disorders. Dystonia, tremor, and parkinsonism were rarely reported, whereas chorea was not reported at all [figure 1]. Seizures were most common in patients with combined myoclonus-ataxia (10/11, 90.9%) and isolated myoclonus (37/48, 77.1%), less frequent in isolated ataxia (8/22, 36.4%), with most myoclonus-ataxia patients exhibiting a progressive myoclonus epilepsy phenotype (10/11, 90.9%) rather than a progressive myoclonus-ataxia syndrome (1/11, 9.1%).
Conclusion: Myoclonus and ataxia are co-occurring movement disorders in GD3 patients, usually associated with seizures, intellectual disability, developmental delay, and oculomotor abnormalities and associated with the “severe” p.Leu483Pro GBA1 variant.
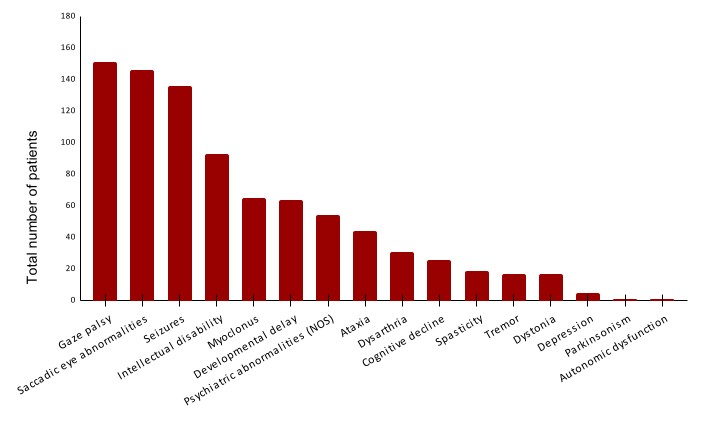
Neurological and psychiatric manifestations
References: 1. Zhong W, Li D, Hong P (2024) A review of type 3 Gaucher disease: unique neurological manifestations and advances in treatment. Acta Neurologica Belgica. 2024;124:1213–1223. https://doi.org/10.1007/s13760-024-02493-1
2. Rossi M, Schaake S, Usnich T et al. (2025) Classification and Genotype–Phenotype Relationships of GBA1 Variants: MDSGene Systematic Review. Mov Disord. 2025 https://doi.org/10.1002/mds.30141
To cite this abstract in AMA style:
S. Schaake, T. Usnich, J. Boehm, N. Steffen, N. Schell, C. Krüger, T. Gül-Demirkale, N. Bahr, T. Kleinz, H. Madoev, B. Laabs, Z. Gan-Or, R. Alcalay, C. Marras, K. Lohmann, C. Klein, M. Rossi. Neurological manifestations and genotypes of Gaucher disease type 3: MDSGene systematic review [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/neurological-manifestations-and-genotypes-of-gaucher-disease-type-3-mdsgene-systematic-review/. Accessed April 10, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/neurological-manifestations-and-genotypes-of-gaucher-disease-type-3-mdsgene-systematic-review/