Category: Ataxia
Objective: This case report discusses the diagnostic difficulties and treatment strategies for Schindler disease type III and provides insightful information on its clinical manifestations
Background: Schindler disease is a rare inherited disorder resulting from a deficiency of α-N-acetylgalactosaminidase activity[1]It is classified into three main phenotypes. Genetic testing(PCR) is the gold standard for diagnosis [2].
Our case correlates with Shindler disease type lll, which makes it our provisional diagnosis with less differential diagnosis like
Autosomal recessive spastic ataxia of Charlevoix-Saguenay
Friedreich Ataxia with Retained Reflexes
Spinocerebellar Ataxia with Axonal
Mitochondrial Recessive Ataxia Syndrome
Our patient was clinically diagnosed; however, due to the rarity of this disease, each new case should be reported. Therefore, genetic testing is highly recommended in our case.
Method: History. A 24-year-old male presented with dysarthri At the age of nine, he developed kinetic tremors in his right hand that progressed to his left hand over a year. By the age of 14, his cognitive and intellectual abilities began to decline. Three years ago developed lower limb weakness and now needs assistance to walk. repetitive vocalizations, talking to himself, laughing and crying simultaneously. He was the first child of consanguineous parents. married with one offspring. He has a 19-year-old younger sister who presents with the same condition as him, with two other normal siblings
Neurological Examination. (MOCA:8), generalized rigidity with cogwheel characteristics, quadriparesis of upper motor neuron pattern, kinetic tremors, dysarthria, diminished bulbar, vocal tics, myoclonic jerks, dystonia. Drunken with high steppage gait. Generalized hyperreflexia with pathological reflexes throughout
Investigations. Vitamin E 9.87, HbA1c 5. Serum lactate 12.43, pyruvate 0.36. MRI brain showed Symmetric ventricular dilation and cortical atrophy.MRI cervical was normal (Figure 1).IQ of 64 (Figure 2). EEG showed bilateral focal epileptogenic activity (Figure 3). Nerve conduction studies and electromyography indicated severe axonal sensorimotor polyneuropathy worsened compared to previous studies (Figure 4).
Results: No specific cure, just symptomatic treatment
Conclusion: No obvious benefit to medications.
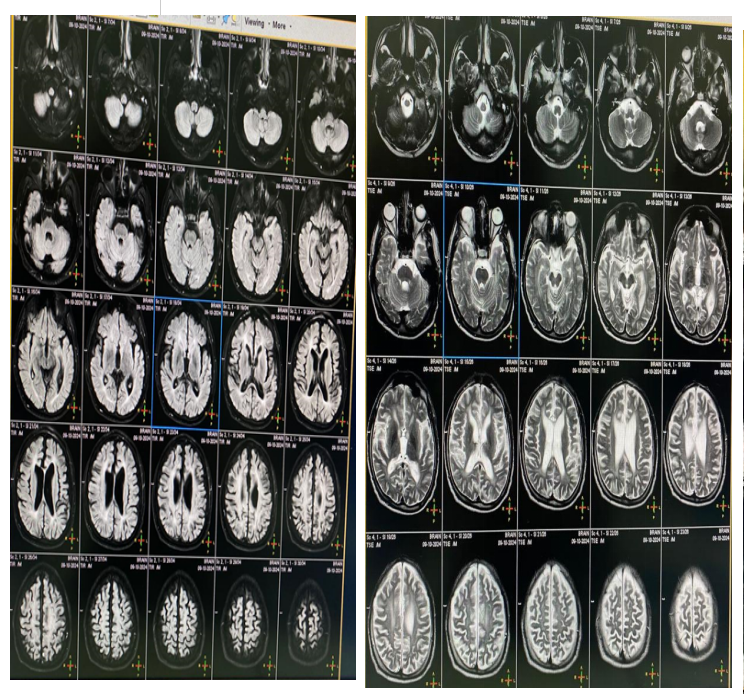
Cortical atrophy

IQ is 64 in Stanford -Binet test
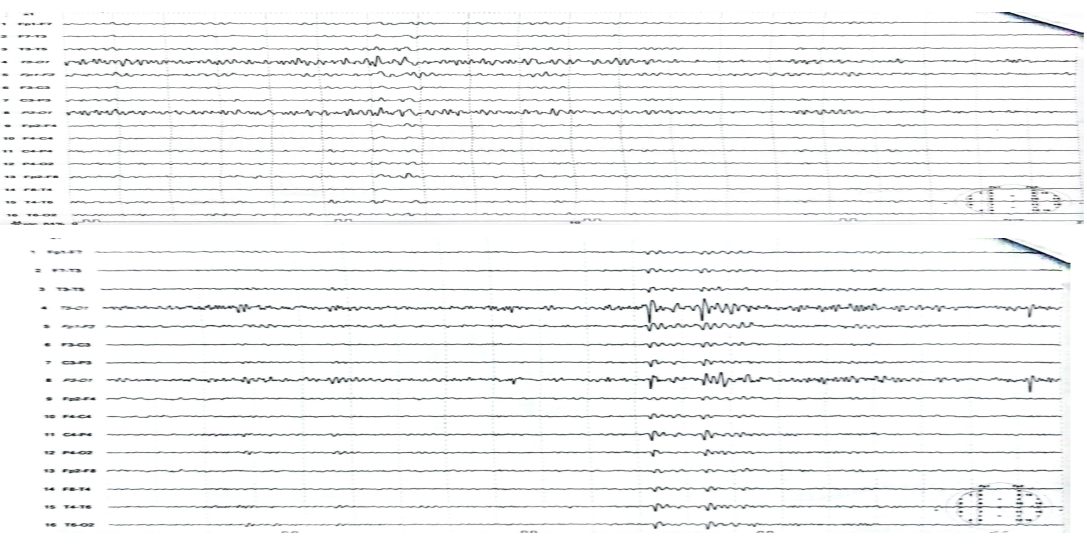
Bilateral focal epileptogenic activity on EEG
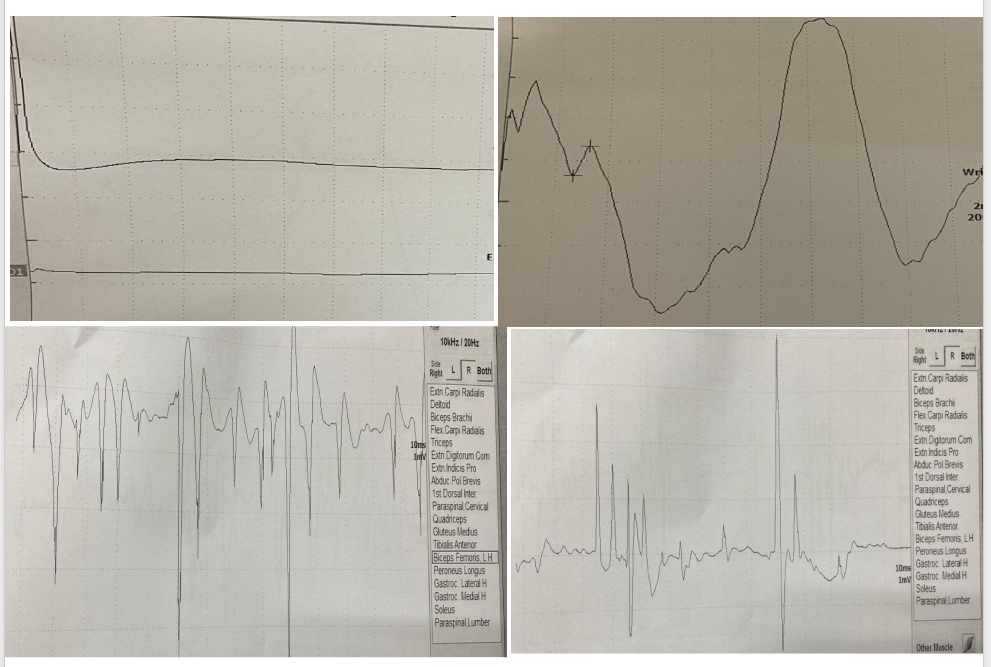
Axonal sensorimotor polyneuropathies affection
References: [1] D. Schindler and R. J. Desnick, “Chapter 49 – Schindler disease: deficient α-N-acetylgalactosaminidase activity,” R. N. Rosenberg and J. M. B. T.-R. M. and G. B. of N. and P. D. (Seventh E. Pascual, Eds., Academic Press, 2025, pp. 709–721. doi: https://doi.org/10.1016/B978-0-443-19041-4.00077-7.
[2] R. G. Castro et al., “A New Case of Schindler Disease.,” 2019, Italy. doi: 10.12890/2019_001269.
To cite this abstract in AMA style:
SH. Elgamal, E. Fahmy, M. Ragab. Schindler Disease (a Rare Autosomal Recessive Lysosomal Storage Disorder) [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/schindler-disease-a-rare-autosomal-recessive-lysosomal-storage-disorder/. Accessed April 7, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/schindler-disease-a-rare-autosomal-recessive-lysosomal-storage-disorder/