Category: Choreas (Non-Huntington's Disease)
Objective: To report a novel stop-gain variant in the RNF213 gene in two individuals, a father and son, presenting with variable neurologic presentations and similar extensive brainstem basal ganglia lesions on brain MRI.
Background: RNF213 (OMIM *613768), is the primary susceptibility gene for moyamoya disease (MMD), a cerebrovascular disorder of the intracranial arteries. More recently, it has been recognized as a Mendelian disease gene, with its phenotypic spectrum expanding to childhood onset stroke-like and encephalopathic presentations, suggesting a broader role in cerebrovascular pathology [1].
Method: Whole exome sequencing (WES) was performed in the proband (Case 1) and his father (Case 2) (Figure 1, Panel A). Repeat expansion analyses for Huntington’s disease, SCA1,2,3,6,7,8,17, and DRPLA were performed in Case 2.
Results: Case 1 presented with childhood onset stroke-like episodes, evolving into an encephalopathic state, and subsequent focal epilepsy. Case 2 presented with adult-onset stroke-like episodes and a Huntington-like phenotype, with progressive generalized chorea, behavioral, and cognitive disturbances. Both subjects presented extensive brainstem and basal ganglia T2/FLAIR hyperintensities on brain MRI (Figure 1, Panel B). Repeat expansion analyses were negative in Case 2. A novel heterozygous STOP-gain variant in RNF213 (NM_001256071.1:c.2157G>A;p.Trp719*) was found in both subjects. The variant is predicted to truncate the protein upstream to the AAA+ adenosine triphosphatase (ATPase) and the Really Interesting New Gene (RING) domains (Figure 1, Panel C). According to the American College of Medical Genetics (ACMG) criteria, this variant was classified as likely pathogenic (PP1, PM2, PVS1, PP3).
Conclusion: In this study, we present a family with two individuals harboring a STOP-gain variant in RNF213 with stroke-like episodes and diffuse brainstem and basal ganglia lesions on neuroimaging, which, in Case 2, are responsible for generalized chorea, behavioral, and cognitive disturbances, mimicking a Huntington’s disease phenotype. These results further validate RNF213 as a Mendelian disease gene and expand its role in cerebrovascular and movement disorders.
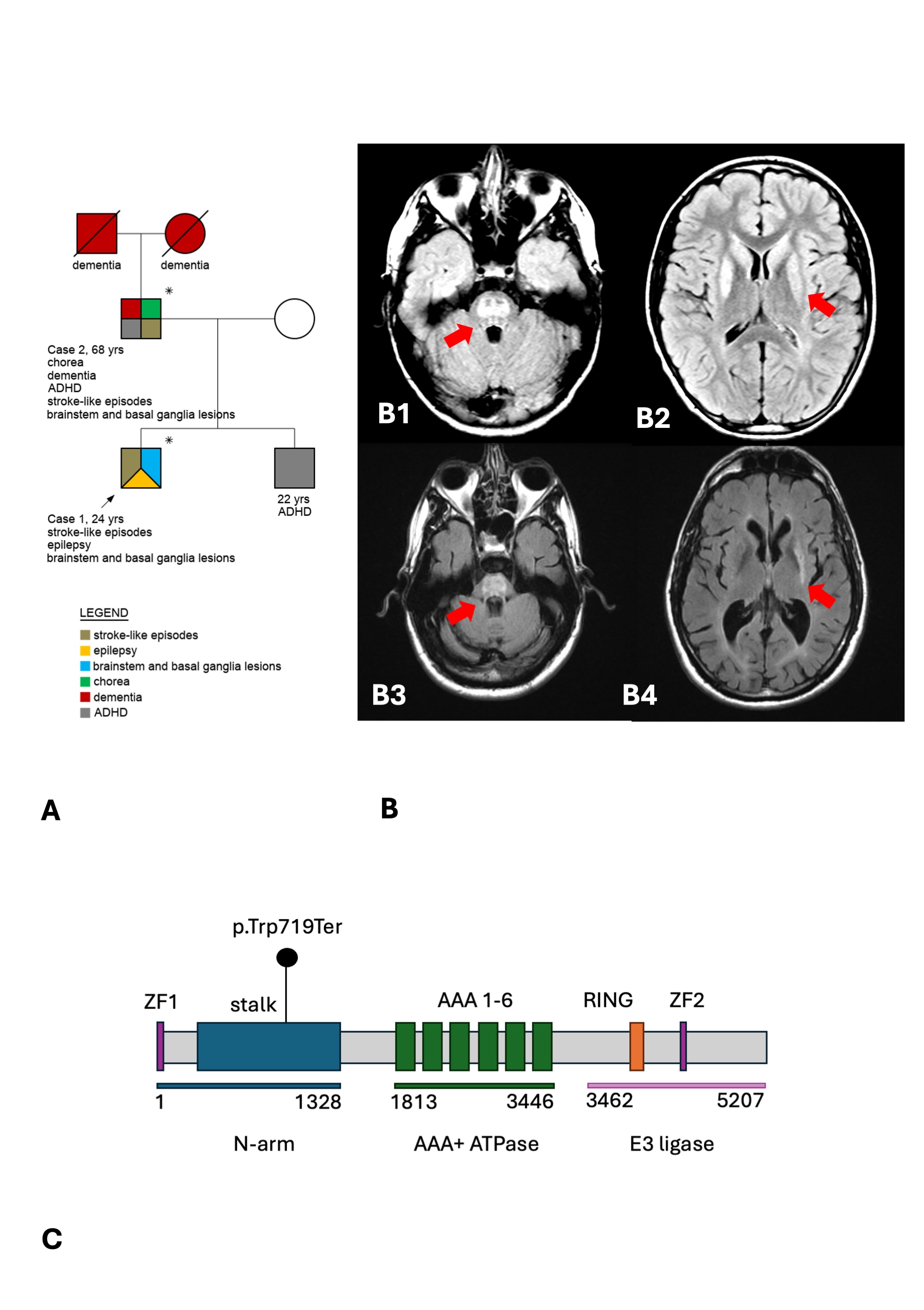
A: family tree. B: brain MRI. C: variant.
References: 1. Brunet T, Zott B, Lieftüchter V, et al (2024) De novo variants in RNF213 are associated with a clinical spectrum ranging from Leigh syndrome to early-onset stroke. Genetics in Medicine 26:. https://doi.org/10.1016/j.gim.2023.101013
To cite this abstract in AMA style:
R. Bovenzi, F. Shen, IJ. Keller Sarmiento, BI. Bustos, L. Kinsley, J. Nichols, D. Krainc, NE. Mencacci. A RNF213 truncating variant causes an autosomal dominant disorder with brainstem and basal ganglia lesions and a Huntington-like phenotype [abstract]. Mov Disord. 2025; 40 (suppl 1). https://www.mdsabstracts.org/abstract/a-rnf213-truncating-variant-causes-an-autosomal-dominant-disorder-with-brainstem-and-basal-ganglia-lesions-and-a-huntington-like-phenotype/. Accessed July 10, 2026.« Back to 2025 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/a-rnf213-truncating-variant-causes-an-autosomal-dominant-disorder-with-brainstem-and-basal-ganglia-lesions-and-a-huntington-like-phenotype/