Session Information
Date: Monday, September 23, 2019
Session Title: Ataxia
Session Time: 1:45pm-3:15pm
Location: Les Muses, Level 3
Objective: To evaluate the clinical manifestation, CAG repeats of abnormal alleles, mitochondrial function and treatment of spinocerebellar ataxia type 6To evaluate the clinical manifestation, CAG repeats of abnormal alleles, mitochondrial function and treatment of spinocerebellar ataxia type 6.
Background: The spinocerebellar ataxias (SCA) is a large group of heterogeneous autosomal dominant degenerative diseases characterized by ataxia, often accompanied by degenerative changes in the brainstem and cerebellum, and other parts of the central nervous system( less commonly the peripheral nervous system, Basal ganglia and thalamus). Among SCA, the spinocerebellar ataxia type 6 accounts for about 15% worldwide, most commonly in Japan, the Netherlands and Germany. In China, there are few case reports and lack of detailed epidemiological data. At present both domestic and international research focus on the gene mutation analysis of SCA6 and there are almost no studies on mitochondrial function. It is significant to study whether there is mitochondrial dysfunction in SCA6, for better understanding of the pathophysiological mechanism and guidance on the treatment in the future.
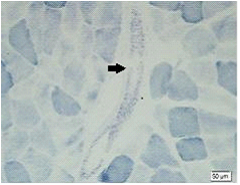
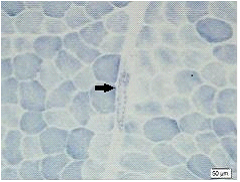
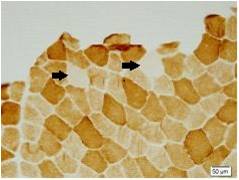
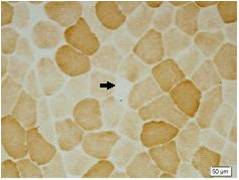
Method: We studied 9 patients from a family with spinocerebellar ataxias. Clinical and molecular features of SCA6 were investigated. We also conducted muscle biopsy to evaluate zitochondrial function. Then 4 patients were treated with Idebenone and Butylphthalide and measured the situation of improvement with scale for the assessment and rating of ataxia scores ( SARA ).
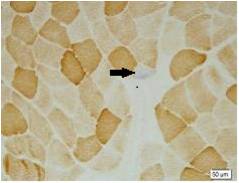
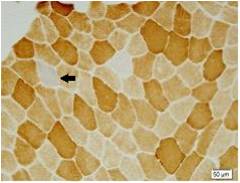
Results: There were totally 9 patients showing slowly progressive ataxia without any remarkable extracerebellar signs in this family and the average age of onset was (40.25±8.76). Brain CT or MRI showed atrophy of the cerebellum. All 4 patients were heterozygous and the number of CAG repeats in the expanded alleles was coincidentally the same 21 repeats and normal alleles in the SCA6 gene ranged from 4–18 CAG units in healthy controls. Muscle biopsy was conducted in 2 patients and it showed mild muscle damage with mitochondrial dysfunction. Idebenone and Butylphthalide are effective in treatment for SCA6.
Conclusion: We observe that CAG expansion in the SCA6 family is completely stable. Clinical features are pure cerebellar impairment and mitochondrial dysfunction. Idebenone and Butylphthalide are effective in treatment for SCA6.
References: [1] Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia associated with small polyglutamine expansions in the á1A-voltage-dependent calcium channel[J]. Nat Genet, 1997, 15:62-69. [2] Ishikawa K, Tanaka H, Saito M, et al. Japanese families with autosomal dominant pure cerebellar ataxia map to chromosome 19p13.1-p13.2, and are strongly associated with mild CAG expansions in the spinocerebellar ataxia type 6 gene in chromosome 19p13.1. Am J Hum Genet[J], 1997, 61: 336-436. [3] Matsuyama Z, Kawakami H, Maruyama H, et al. Molecular features of the CAG repeats of spinocerebellar ataxia 6 (SCA6)[J]. Hum Mol Genet, 1997, 6:1283-1287. [4] Reiss O, Schöls L, Böttger H, et al. SCA6 is caused by moderate CAG expansion in the á1A-voltage-dependent calcium channel gene[J]. Hum Mol Genet, 1997, 6: 1289–1293. [5] Soong B, Liu R, Wu L, et al. Metabolic characterization of spinocerebellar ataxia type 6[J]. Arch Neurol, 2001, 58: 300-304. [6] Ikeuchi T, Takano H, Koide R, et al. Spinocerebellar ataxia type 6: CAG repeat expansion in alpha1A voltage-dependent calcium channel gene and clinical variations in Japanese population[J]. Ann Neurol, 1997, 42: 879-884. [7] Geschwind DH, Perlman S, Figueroa KP, et al. Spinocerebellar ataxia type 6 Frequency of the mutationand genotype-phenotype correlations[J]. Neurology, 1997, 49: 1247-1251. [8] van de Warrenburg BP, Hendriks H, Durr A, et al. Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch-French cohort[J]. Ann Neurol, 2005, 57: 505-512. [9] Gomez CM, Thompson RM, Gammack JT, et al. Spinocerebellar ataxia type 6: gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset[J]. Ann Neurol, 1997, 42: 933-950. [10] Ishikawa K, Watanabe M, Yoshizawa K, et al. Clinical, neuropathological, and molecular study in two families with spinocerebellar ataxia type 6[J]. J Neurol Neurosurg Psychiatry, 1999, 67: 86-89. [11] 王雪晶,沈若武,黄良,等. 早发型脊髓小脑共济失调6型家系的神经病理学研究[J]. 中华神经科杂志, 2009, 42: 509-513.
To cite this abstract in AMA style:
H. Luan, A. Liu. Clinical and molecular features of a Chinese family with spinocerebellar ataxia type 6 [abstract]. Mov Disord. 2019; 34 (suppl 2). https://www.mdsabstracts.org/abstract/clinical-and-molecular-features-of-a-chinese-family-with-spinocerebellar-ataxia-type-6/. Accessed August 7, 2026.« Back to 2019 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/clinical-and-molecular-features-of-a-chinese-family-with-spinocerebellar-ataxia-type-6/