Session Information
Date: Saturday, October 6, 2018
Session Title: Genetics (Non-PD)
Session Time: 1:45pm-3:15pm
Location: Hall 3FG
Objective: We present a case series of adult onset ataxia with variability in genetic mutations, yet common clinical phenotypic presentations that constitute the autosomal recessive form of spinocerebellar ataxia (SCA).
Background: Hereditary ataxias are classified as autosomal dominant, autosomal recessive, X-linked and mitochondrial. Despite the differences in presentation, these ataxias share a clinical phenotypic presentation of impaired and unsteady ambulation, changes and decline in balance, and a lack of coordination of gait and limbs. Additional clinical symptoms include pyramidal features, peripheral neuropathy, extrapyramidal signs, cognitive loss and/or retinopathy. Well known forms of autosomal recessive cerebellar ataxias (ARCAs) in Caucasians include Friedreich ataxia, accounting for 30–40% of patients with ARCA in the adult population. Other less frequent ARCAs found in the adult population include autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS), ataxia with vitamin E deficiency (AVED), and spinocerebellar ataxia with neuropathy 1 (SCAN1). Our patients, in addition to demonstrating mutations in SYNE1, demonstrate other genetic mutations which may impact presentation and progression.
Methods: A case series of 5 patients with the chief complaint of gait ataxia were assessed in an outpatient Movement Disorders specialty clinic, with clinical confirmation. MRI imaging results consistent with cerebellar atrophy, lead to obtaining genetic panels for SCA. Patients were followed clinically over a range of 2-5 years for progression, or change in clinical presentation.
Results: The clinical phenotypic, radiologic and genetic findings of the 5 patients are shown in Tables 1 and 2.
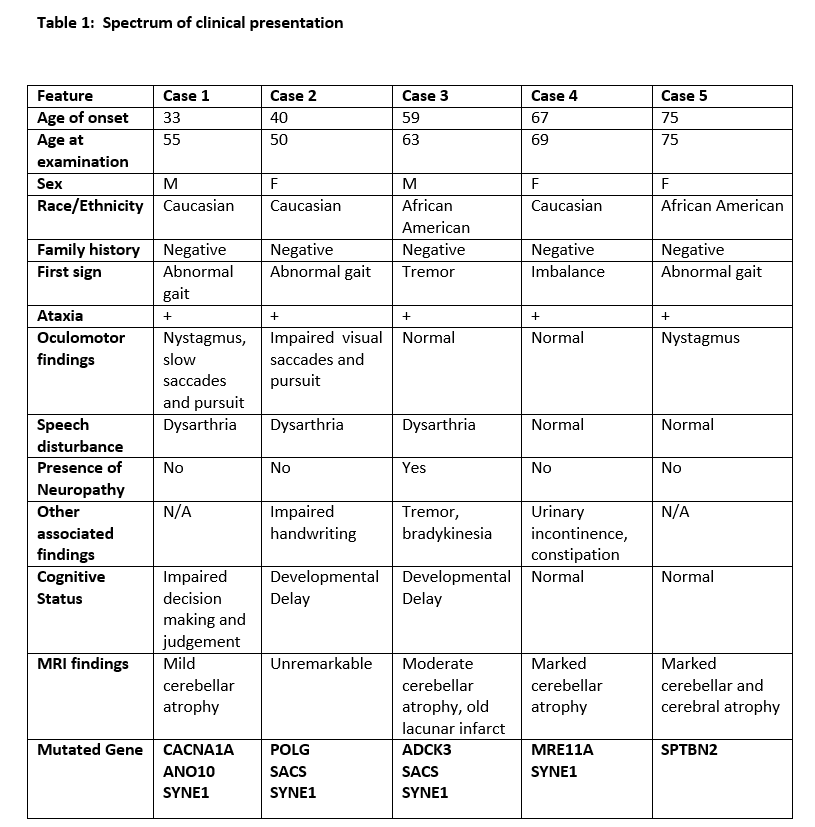
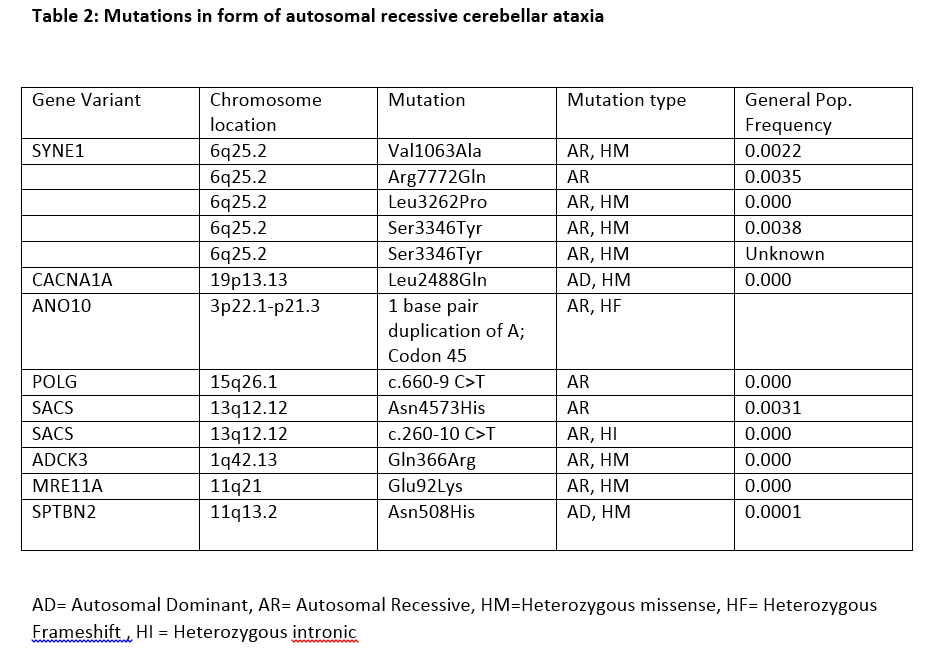
Conclusions: The genetic observations in this case series suggests a pattern of phenotypic presentation that is likely associated with at least one copy of SYNE1, however, this is unlikely to be the independent biological etiology. Most of our patients had more than one mutation, perhaps pointing to an impact on their core phenotypic presentation and disease progression of mutation burden. Overall, we report an ARCA phenotype in accordance with previous literature with phenotypic definition as outlined, which becomes helpful when facing a patient with recessive or sporadic ataxia. Further research into autosomal recessive SCA is needed to delineate the phenotypic presentation, genetic composition, and prognosis for these patients.
References: Gros-Louis F, Dupré N, Dion P, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat. Genet. 2006;39:80.
To cite this abstract in AMA style:
A. Persaud, O. Oguh. Expanding the clinical phenotype of autosomal recessive spinocerebellar ataxia with Adult onset; a collection of case series [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/expanding-the-clinical-phenotype-of-autosomal-recessive-spinocerebellar-ataxia-with-adult-onset-a-collection-of-case-series/. Accessed August 10, 2026.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/expanding-the-clinical-phenotype-of-autosomal-recessive-spinocerebellar-ataxia-with-adult-onset-a-collection-of-case-series/