Session Information
Date: Saturday, October 6, 2018
Session Title: Rare Genetic and Metabolic Diseases
Session Time: 1:45pm-3:15pm
Location: Hall 3FG
Objective: To describe an early onset movement disorder due to impairment of valine metabolism.
Background: HIBCH deficiency is a rare inborn error of valine metabolism identified in 1982 [figure1]. However, most cases so far reported were diagnosed in the last few years by NGS [table1-2]. We report on a new case with a prolonged follow-up diagnosed on the basis of a chemical biomarker.
Methods: Case report.
Results: This 34-year-old man showed motor skill developmental delay and recurrent attacks of chorea during febrile illnesses since the age of 8 months. He walked unsupported with an ataxic gait at the age of 2 while other psychomotor milestones were reported normal until the age of 6, when he experienced neurological regression and epileptic seizures during a febrile disease. On examination he showed severe hypotonia and hypokinesia with loss of postural reactions and cerebellar dysarthria. Partial neurological recovery was observed in the following months. At the age of 9 he presented an ataxic-dyskinetic-spastic gait with distal dystonic movement and posturing of upper limbs. On the last examination, at the age 34, he was wheelchaired and showed moderate to severe intellectual disability, spastic-dystonic tetraparesis, severe hypo- and bradykinesia, head titubation, ophthalmoplegia, optic atrophy and retinopathy, lower limb axonal peripheral neuropathy. Serial MRI disclosed bilateral pallidum alteration on T2 sequences and cerebral atrophy (Fig.2). 123I-ioflupane brain SPECT and H-MRS were normal. Mitochondrial respiratory chain on a muscle biopsy was unaffected. A dosage of acylcarnitine on dried blood spot (DBS) showed high levels of 3-idroxybutyrylcarnitine (C4OH) and Sanger sequence of HIBCH gene revealed a novel homozygous variant (c.777T>A, p.Phe259Leu).
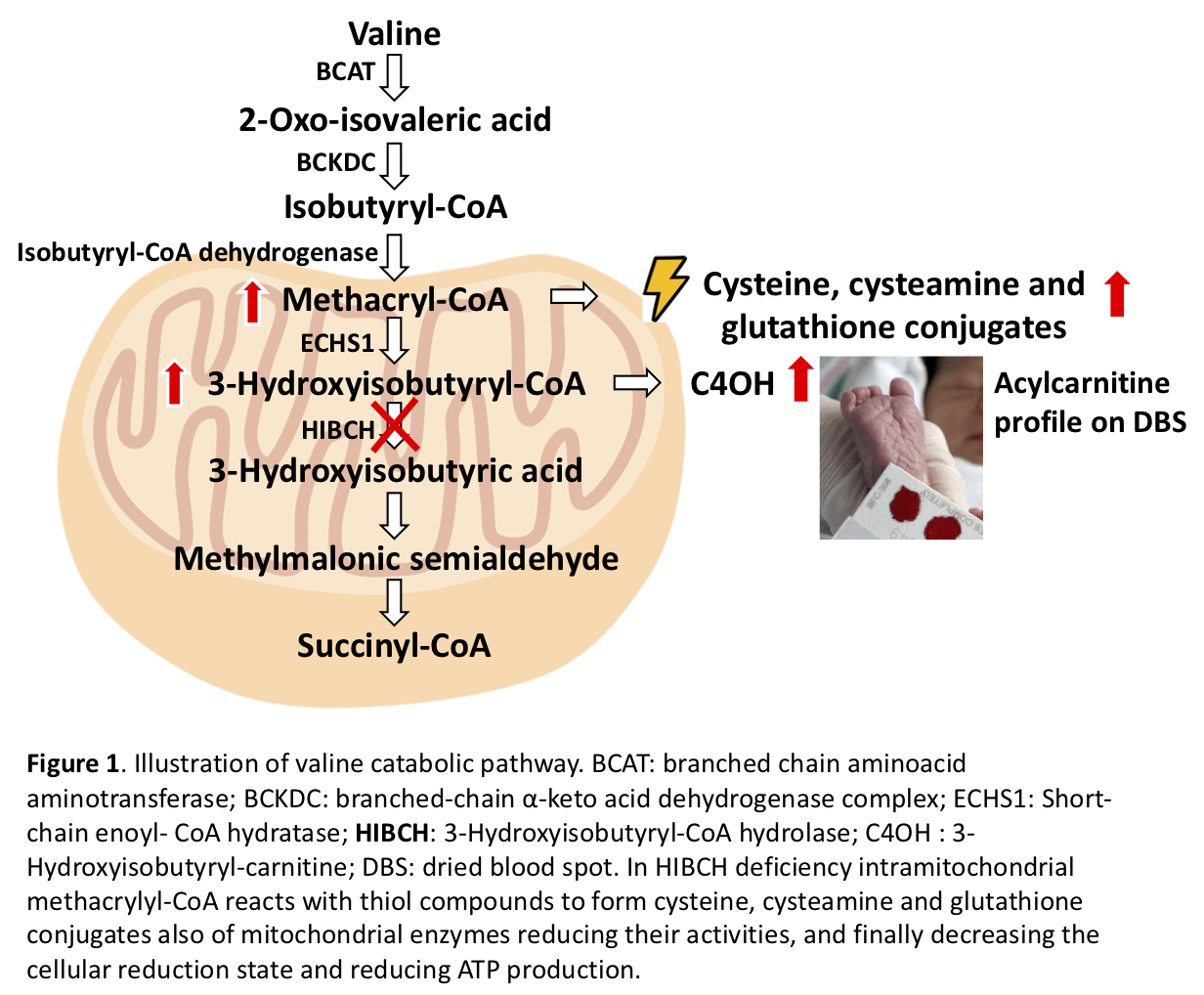
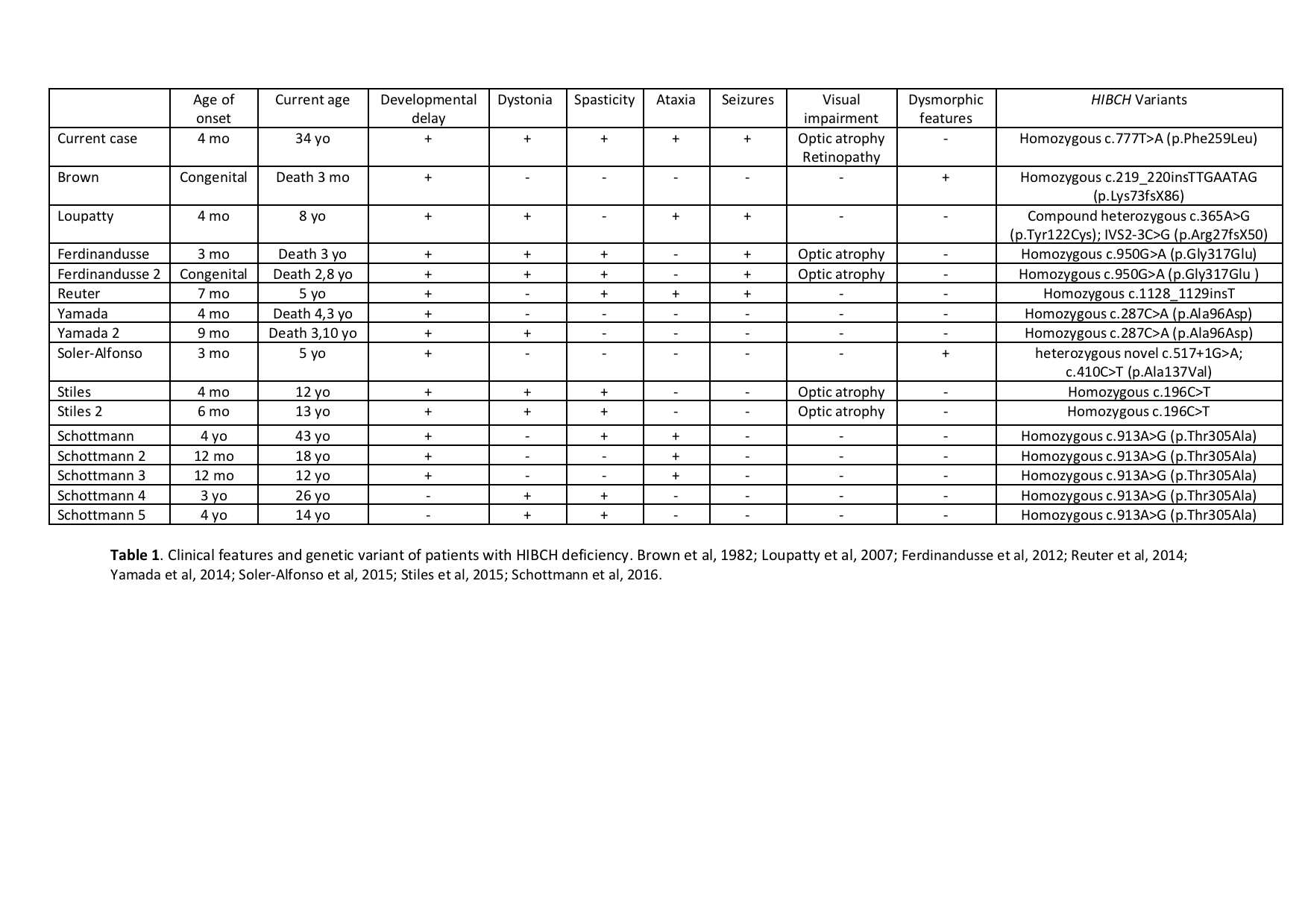
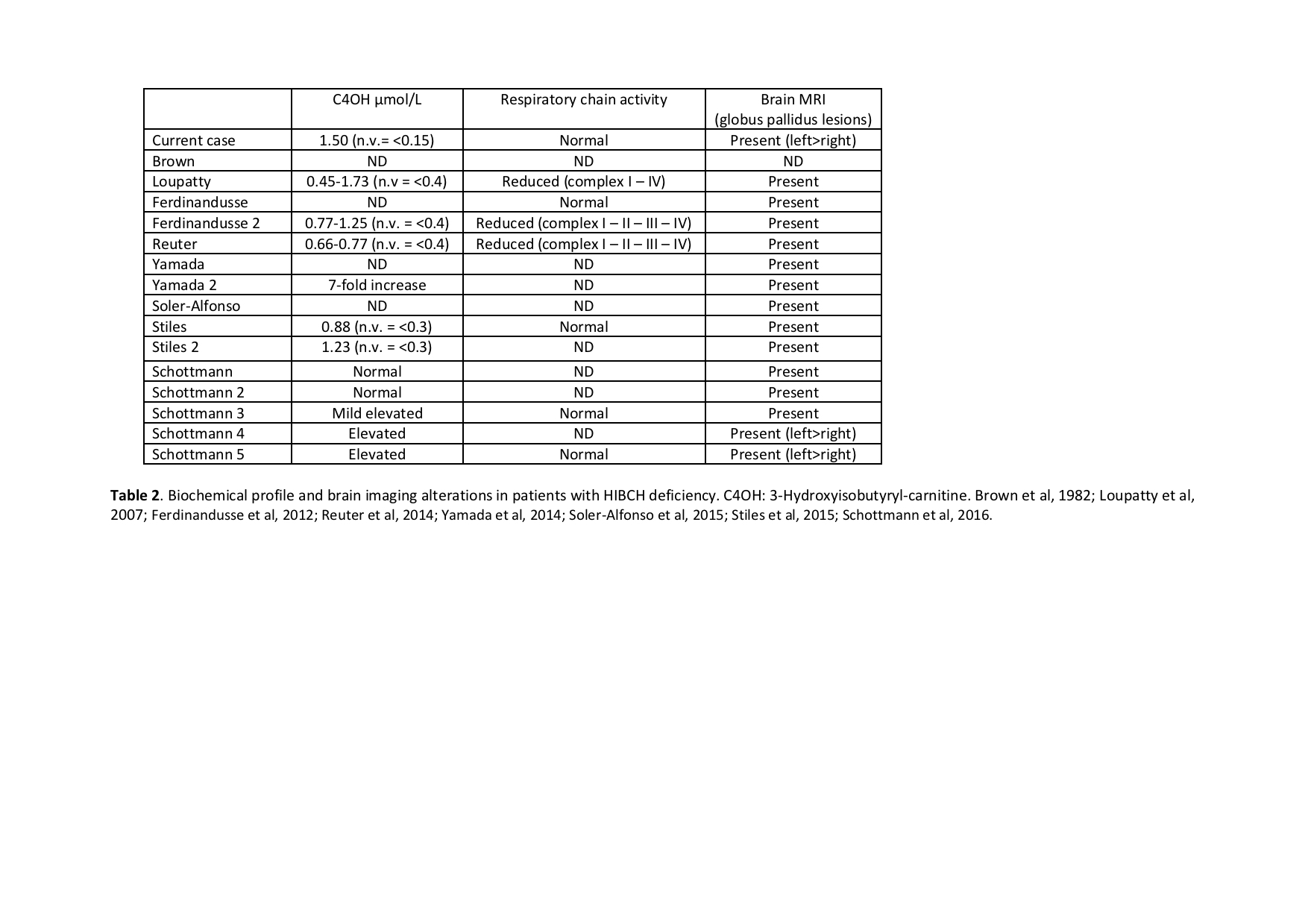
Conclusions: HIBCH defect is an underestimated cause of early onset and slowly progressive Leigh syndrome due to a secondary damage of mitochondrial function. The diagnosis may be promptly performed by the dosage of acylcarnitine on DBS, possibly as part of neonatal screening program. If early diagnosed the disorder is potentially treatable by handling valine intake with the diet and by contrasting oxidative damage. With respect to the few reported cases our patient developed peripheral neuropathy and retinopathy. Finally, isolated pallidal lesion may be suggested as a neuroimaging diagnostic marker of this condition.
References: Schottmann G, Sarpong A, Lorenz C, Weinhold N, Gill E, Teschner L, Ferdinandusse S, Wanders RJ, Prigione A, Schuelke M. A movement disorder with dystonia and ataxia caused by a mutation in the HIBCH gene. Mov Disord. 2016 Nov;31(11):1733-1739. Yamada K, Naiki M, Hoshino S, Kitaura Y, Kondo Y, Nomura N, Kimura R, Fukushi D, Yamada Y, Shimozawa N, Yamaguchi S, Shimomura Y, Miura K, Wakamatsu N. Clinical and biochemical characterization of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency that causes Leigh-like disease and ketoacidosis. Mol Genet Metab Rep. 2014 Oct 16;1:455-460.
To cite this abstract in AMA style:
F. Nardecchia, L. Pollini, C. Carducci, M. Tolve, S. De Leo, C. Carducci, V. Leuzzi. Movement disorder associated with 3-Idroxyisobutyryl-Coa hydrolase (HIBCH) deficiency [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/movement-disorder-associated-with-3-idroxyisobutyryl-coa-hydrolase-hibch-deficiency/. Accessed August 6, 2026.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/movement-disorder-associated-with-3-idroxyisobutyryl-coa-hydrolase-hibch-deficiency/