Category: Rare Genetic and Metabolic Diseases
Objective: To describe 5 late-onset sporadic cases carrying either biallelic or monoallelic variants in ATP13A2, showing either spastic ataxia(Spatax) or primary lateral sclerosis (PLS) as core phenotype.
Background: ATP13A2-PARK 9 is a lysosomal transport-ATPase, with a pivotal role in both lysosomal function and mitochondrial network integrity. Biallelic pathogenic variants in ATP13A2 gene have been associated to Kufor-Rakeb disease, ALS, HSP, and Neuronal Ceroid Lipofuscinosis1. More recently, also a possible causative significance for monoallelic variants has been proposed, specifically as a risk factor for PD and Multiple System Atrophy2.
Method: Two Spatax patients resulted negative for FXN, FMR1 and CAG expansion-associated cerebellar ataxias and three patients with upper motor neuron features underwent next-generation-sequencing targeted resequencing panels for ataxia and motor neuron disease, respectively.
Results: Spatax cases comprised two females with late-onset (63 and 52 years, respectively) gait difficulties. Both of them showed cerebellar and spastic signs, with more severe spasticity in Pt1, who was wheelchair-bound after 7 years from onset. Brain MRI showed cerebellar atrophy in Pt1, while was normal in Pt2. Pt1 carried the monoallelic p.Gln382Ter variant in ATP13A2, while Pt2 had the p.Met766Val variant. Pt3-5 had upper motoneuron phenotype, with mean age at onset of gait difficulties at 57 years. Pt3 was a 72-year female showing PLS phenotype with mixed cerebellar-spastic dysarthria and cerebellar atrophy at brain MRI(figure). Pt4 was a 56-year male with PLS and nystagmus, mixed cerebellar and spastic dysarthria and white matter abnormalities (WMA) at brain MRI, evolving to amyotrophic lateral sclerosis (ALS) in about 6 years. The last case, Pt5, was a 73-year female with HSP phenotype at onset, evolving to ALS after about 5 years from onset, with WMA at brain MRI. Except for the p.Gln382Ter, which was predicted to be pathogenic, all the other variants were classified as variants of uncertain significance (VoUS).
Conclusion: In our 2 heterozigous patients both the late-onset of symptoms and the phenotype, being similar to ATP13A2 related AR neurological diseases might support a potential causative role of the VoUS. Functional studies on fibroblast-patient derived cells are needed to address this issue. If confirmed, this will expand spectrum of ATP13A2 variants associated phenotypes.
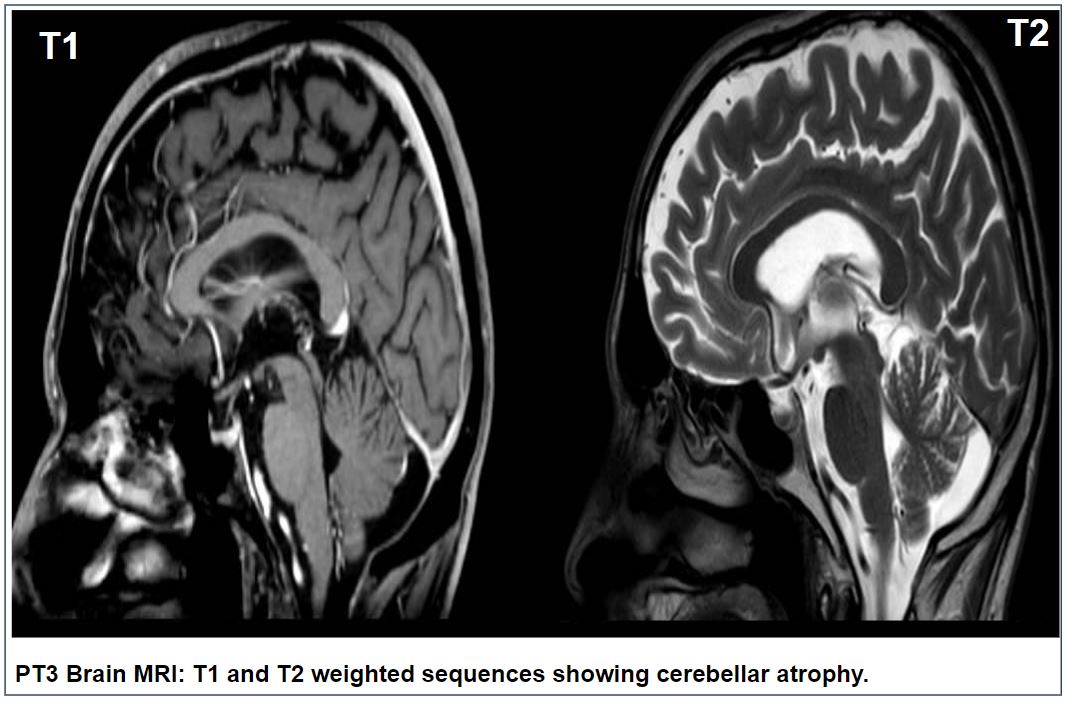
PT3 Brain MRI: showing cerebellar atrophy.
References: 1) Yang X, Xu Y. Mutations in the ATP13A2 gene and Parkinsonism: a preliminary review. Biomed Res Int. 2014;2014:371256. doi:10.1155/2014/371256. Epub 2014 Aug 14. PMID: 25197640; PMCID: PMC4147200.
2) Park JS, Koentjoro B, Klein C, Sue CM. Single Heterozygous ATP13A2 Mutations Cause Cellular Dysfunction Associated with Parkinson’s Disease. Mov Disord. 2018 May;33(5):852-854. doi: 10.1002/mds.27327. Epub 2018 Feb 14. PMID: 29442384.
To cite this abstract in AMA style:
G. Dalla Zanna, A. Funcis, S. Rossi, F. Santorelli, M. Sabatelli, G. Silvestri. Spastic Ataxia and Motorneuron disease as possible manifestations of ATP13A2 (PARK 9) variants [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/spastic-ataxia-and-motorneuron-disease-as-possible-manifestations-of-atp13a2-park-9-variants/. Accessed June 22, 2026.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/spastic-ataxia-and-motorneuron-disease-as-possible-manifestations-of-atp13a2-park-9-variants/