Objective: Uncover novel molecular signatures to define Parkinson’s disease (PD) patients’ clusters using multi-omics integration and evaluate clinical and neuroimaging data.
Background: PD is a heterogeneous multifactorial neurodegenerative disorder with identifiable clinical-pathological subtypes. A comprehensive molecular profile could reduce their phenotypic heterogeneity. We integrated multi-omic data from the Parkinson’s Progression Markers Initiative cohort to identify molecularly defined PD subclusters.
Method: We selected PD patients (n = 608), including LRRK2 (n = 155) and GBA (n = 76) mutation carriers and sporadic PD (sPD) cases (n = 377) with cerebrospinal fluid (CSF) and urine proteomics, blood transcriptomics, and plasma metabolomics, and the expression data were log2-transformed and normalized, and features with > 20% missing values were excluded. Integrative clustering across multi-omics using the top 2000 most-variable proteins and transcripts was performed using iClusterBayes[1] to cluster patients with PD and informative molecular features. We tested the differences across clinical assessments by ordinal logistic regression for ordinal outcomes (Gastrointestinal (GI) severity, cognitive and motor scores) and t-test for the continuous outcome (DaTscan volumetric data, CSF synuclein (αSyn) levels), and logistic regression (αSyn seed amplification assay (αSyn-SAA)). We applied a linear mixed model to capture longitudinal heterogeneity of clinical progression between PD subtypes.
Results: We uncovered two multi-omic clusters (n1 = 153 and n2 = 220) determined by 11 CSF and 1082 urine proteins and six blood transcripts that exhibit different proportions of mutation types (p = 4.5×10-5), disease duration (p = 2.2×10-7), and UPDRS-II scores (p = 0.03) [figure 1-2]. Cluster 1 has more LRRK2 carriers, showing faster progression in UPDRS-II [figure 3], and displays lower levels of a metabolite, lysophosphatidylethanolamine (LPE) (log2FC = -0.22, p = 0.0013) [figure 4]. A model with αSyn-SAA, DaTscan, and CSF αSyn can distinguish PD from controls (AUC = 0.95) [figure 5]. We uncovered a CSF αSyn-SAA genetic modifier (OR = 0.32, p = 6.2 x10-8) in chromosome 11 [Figure 6].
Conclusion: Integrating multi-omics data from PD patients provides insights into the molecular mechanisms and clinical significance within distinct PD subclusters.
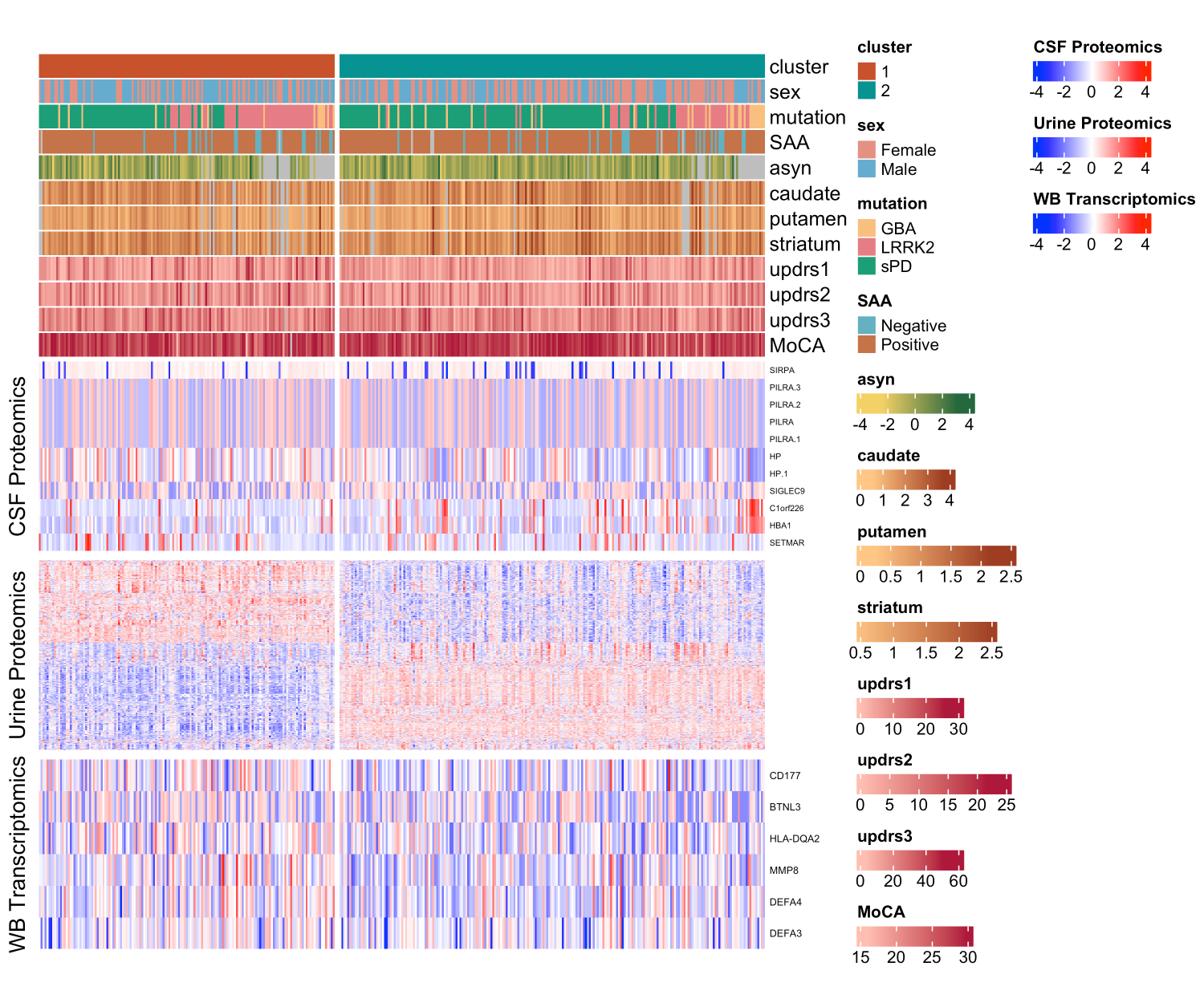
Heatmap of PD clustering
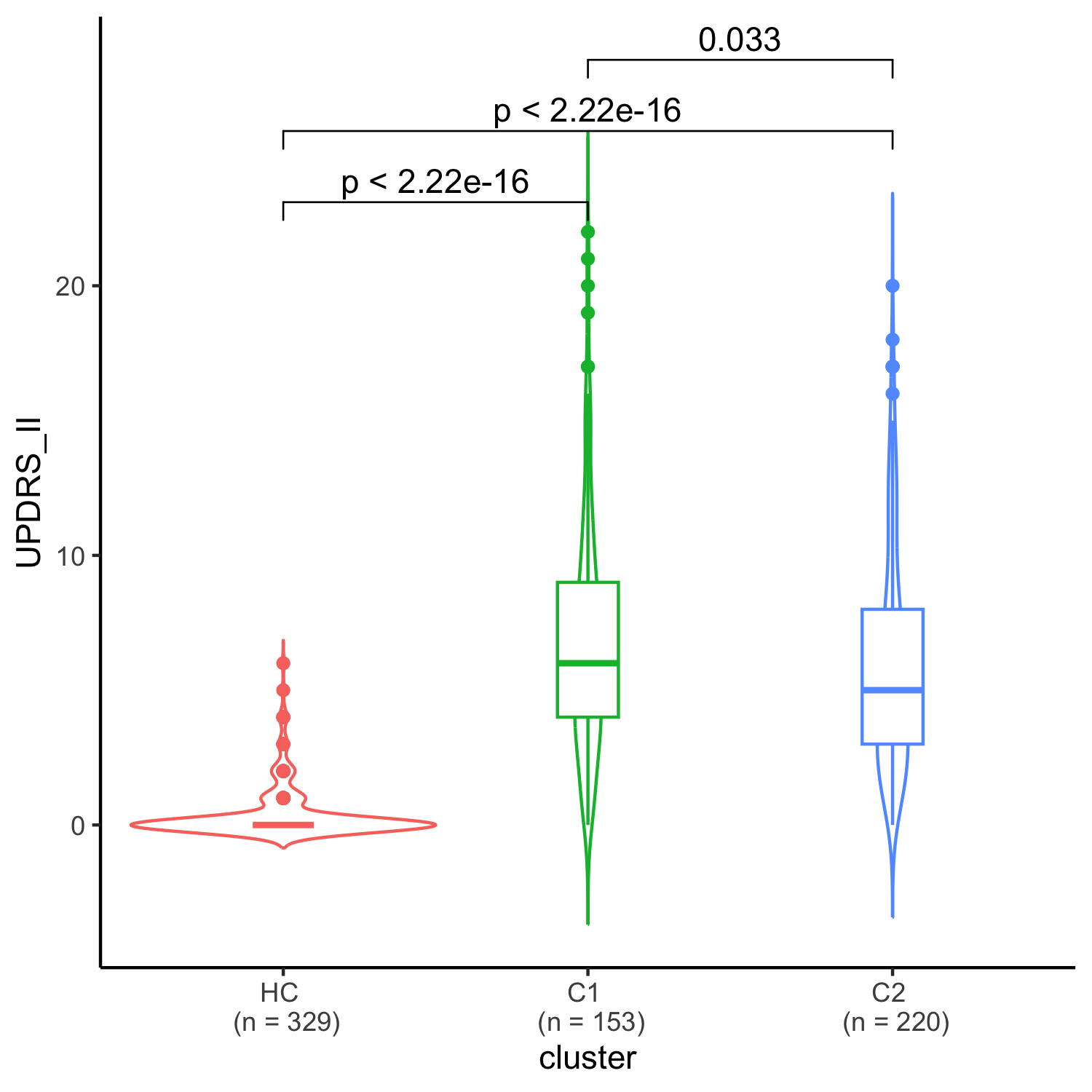
UPDRS II scale comparisons
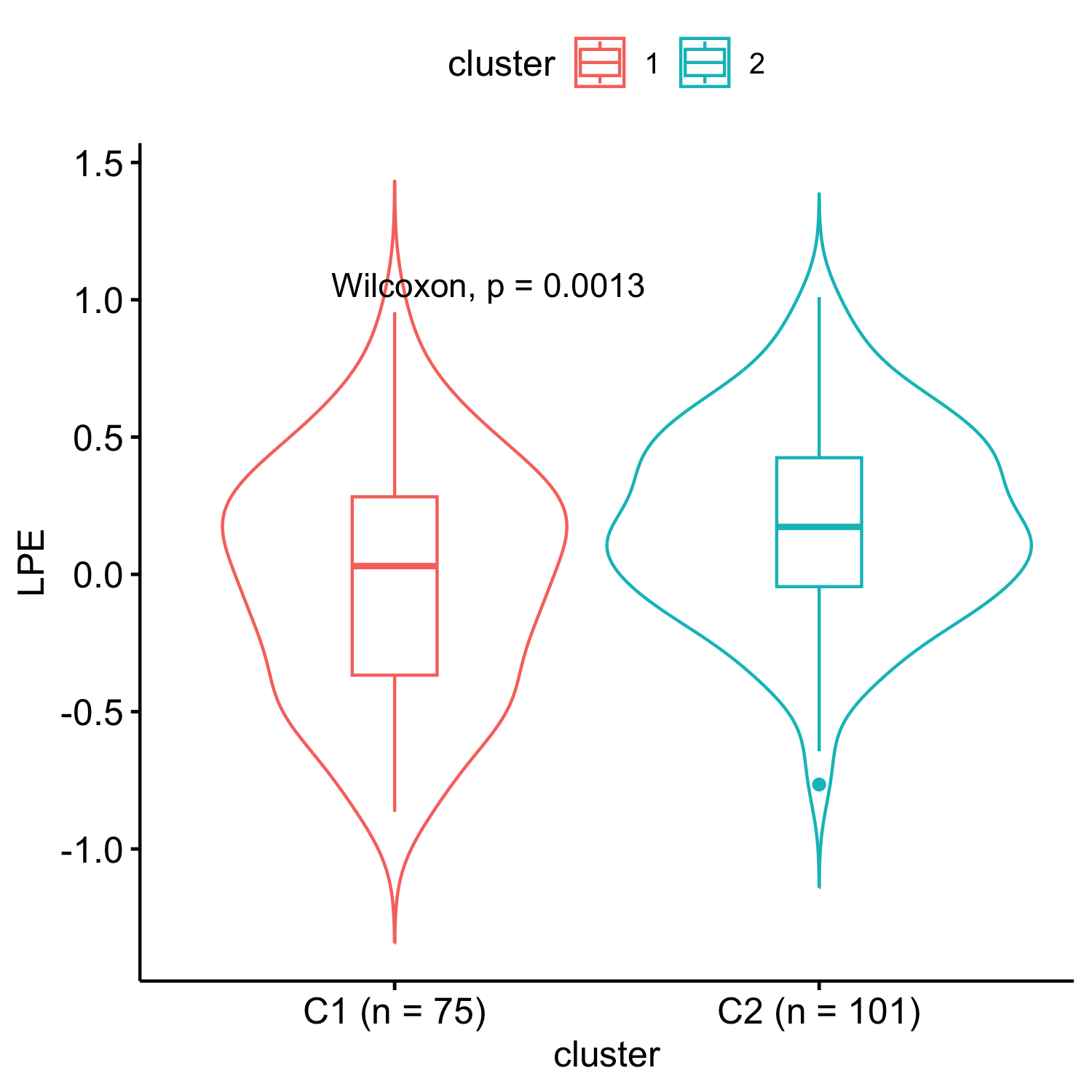
LPE levels comparison
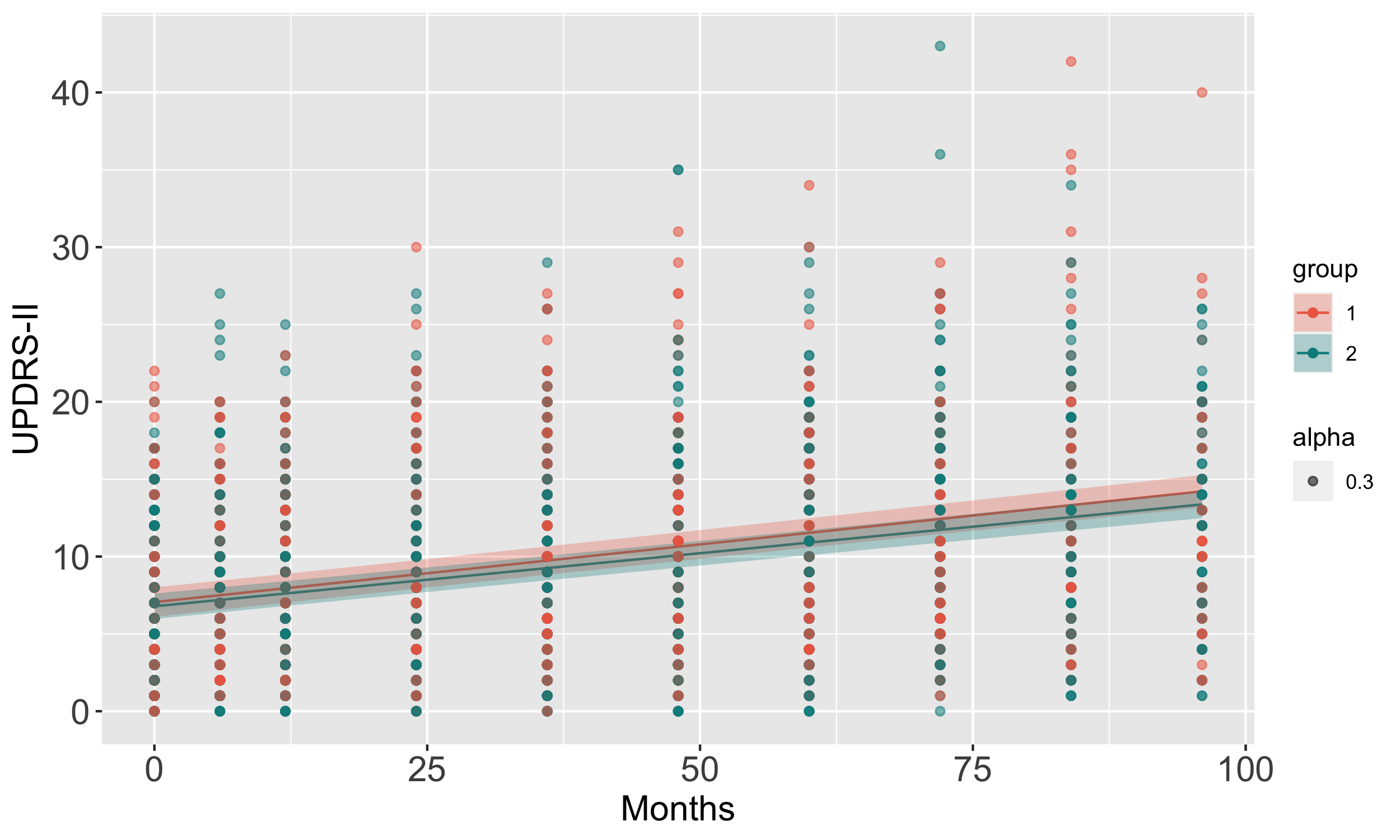
UPDRS-II trajectories over five years
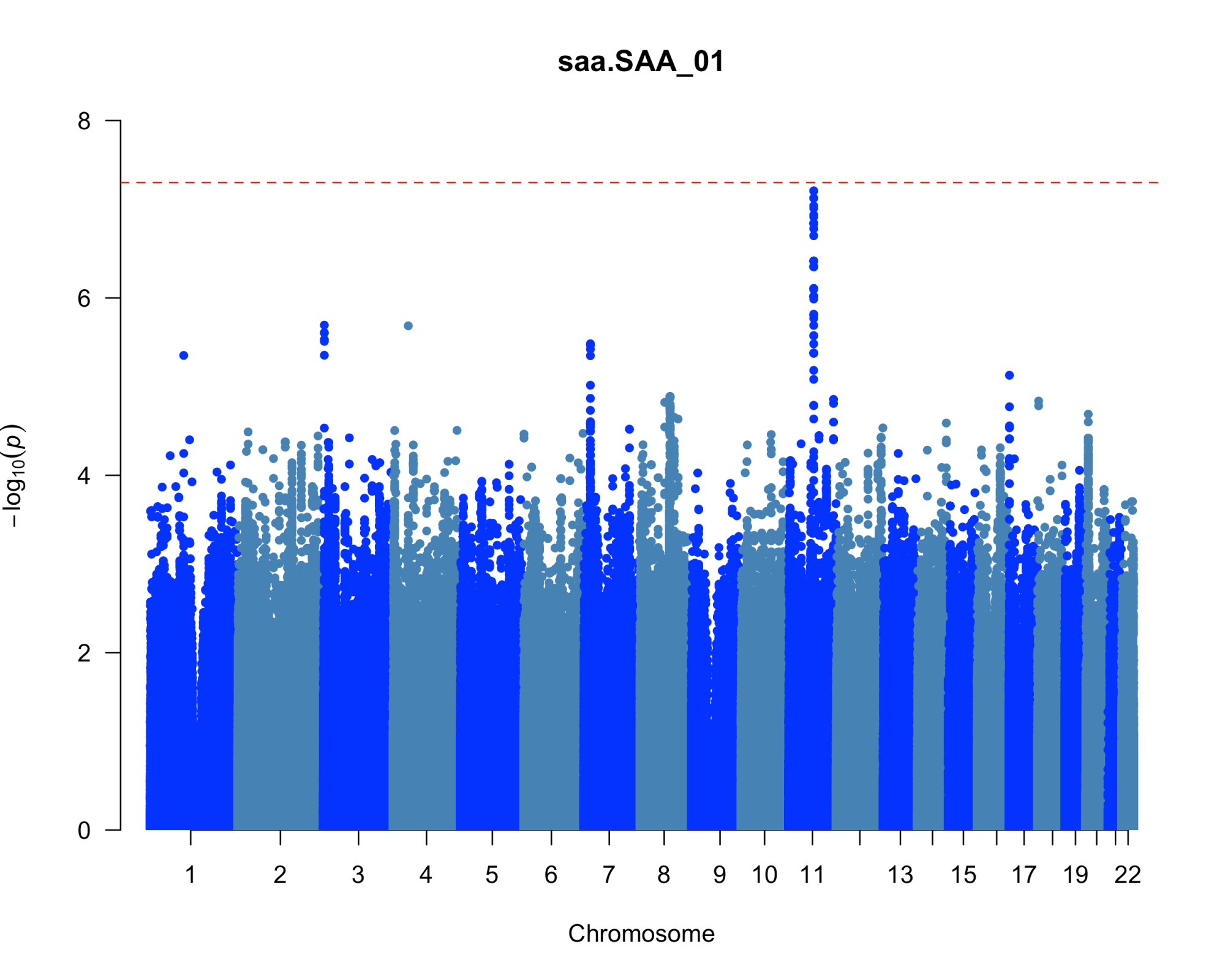
Manhattan plot of GWAS for SAA
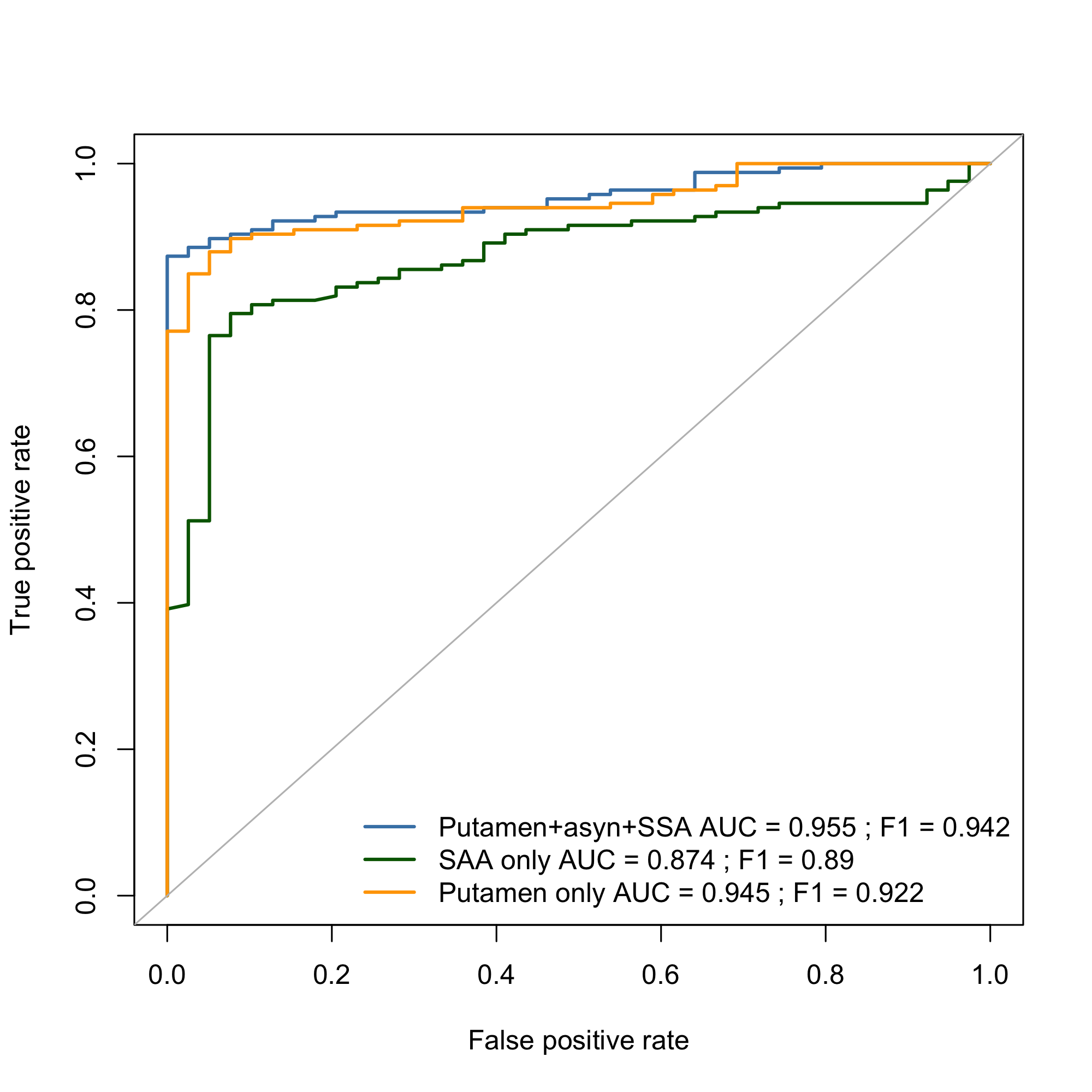
ROC curves
References: 1. Mo, Q., Shen, R., Guo, C., Vannucci, M., Chan, K. S., & Hilsenbeck, S. G. (2018). A fully Bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics, 19(1), 71-86.
To cite this abstract in AMA style:
M. Lai, B. Benitez. Cross-Omics Clustering Identifies Common Molecular Patterns in Parkinson’s Disease [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/cross-omics-clustering-identifies-common-molecular-patterns-in-parkinsons-disease/. Accessed August 5, 2026.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/cross-omics-clustering-identifies-common-molecular-patterns-in-parkinsons-disease/